| Pages:
1
..
18
19
20
21
22
..
33 |
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Evidence has been found that establishes to a virtual 100% certainty that my earlier suspicions and conclusions are correct that both Von Herz and
Klapotke have misidentified the DDNR compound as 6-diazo when it is actually a 4-diazo.
I have referenced the original work of Fitz regarding the dinitroso derivative of resorcinol which is 2,4-dinitroso resorcinol confirmed by later
reviewers.
My earlier assertion about the name for the compound has gained support
4-diazo-2,6-dinitroresorcinol-(1-anhydride) * most likely structure IMO
or for the alternative hydroxyl location for the anhydride Oxygen if that proves to be the case
4-diazo-2,6-dinitroresorcinol-(3-anhydride)
See attached articles and this linked article preview
http://pubs.acs.org/doi/abs/10.1021/jo00258a039
which identifies the much different conditions required for producing the 4,6-dinitroresorcinol which is a historically much later method than the
methods of Benedikt and Hubl derived from Fitz, and later repeated by Von Herz, most recently by Klapotke using the different approach of working as
did Meldola and Reverdin with nitrated paracetamol derivatives.
The mono amido derivative of 2,4-dinitroresorcinol and the mono amido derivative of 2,4,6-trinitroresorcinol (styphnamic acid) both lead to DDNR.
6-amino cannot be present upon the mono amido derivative of 2,4-dinitroresorcinol, which excludes the 6 position for the resulting diazo derivative
DDNR, barring any unlikely and improbable diazo rearrangement.
This leaves position 4 as the most likely amino location for both the mono amido derivative of 2,4-dinitroresorcinol and styphnamic acid.
The remaining possibility of a 2 nitro / 2 amino becoming a 2 diazo seems unlikely, but even at that would seem more possible than a 6-diazo which IMO
is just plain wrong. I think the possibility of a 2 nitro / 2 amino has already been ruled out in the early literature, but I don't have the
citation.
Anyway, I am sticking to my guns that Von Herz GB207563 and Klapotke are both wrong about the 6 diazo structure which is in nearly all probability a
4-diazo on DDNR.
The synthetic approach of Benedikt and Hubl, Von Herz, et al to DDNR appears to be a technically superior method working from resorcinol itself as a
starting material, compared with the alternative approach from paracetamol. All things considered, the use of styphnamic acid as the precursor for
DDNR would seem the least technically complicated. The styphnic acid precursor could be reduced to styphnamic acid in the same way as is made
picramic acid and the diazotization could then be done by the method of Benedikt and Hubl (and Von Herz) or in the alternative by the method of Hagel
and Redecker US4246052. I concur with the structural identification of Hagel and Redecker and I disagree with the structural identification of Von
Herz and Klapotke.
It is also possible that the 2,4-dinitroresorcinol may be subjected to reduction to 4-amino-2-nitroresorcinol and then diazotized by the method of
Benedikt and Hubl to form DDNR.
Patents of interest for 2,4-dinitroresorcinol are US2945890 and US2811565 and US3933926. Further nitration to styphnic acid via a dinitroso
intermediate see US2301912.
Both approaches to DDNR are technically feasible but the approach from resorcinol involves fewer and less complicated synthetic steps and less loss
from manipulations of unstable intermediates. However the paracetamol route is necessary for the related compound
2,3,6-trinitro-4-diazophenol-(1-anhydride) which is a potentially useful energetic compound, but will likely have inadequate stability to be
practical.
Attachment: Fitz Berichte 1875.pdf (317kB)
This file has been downloaded 698 times
Attachment: US2945890 2,4-dinitroresorcinol.pdf (158kB)
This file has been downloaded 604 times
Attachment: US2811565 2,4-dinitroresorcinol.pdf (306kB)
This file has been downloaded 890 times
Attachment: Dinitrosoresorcinol Fitz A_Dictionary_of_Chemistry_pg1750.pdf (289kB)
This file has been downloaded 821 times
Attachment: DinitrosoResorcinol JACS 1923, 45, pg 1536 to 1539.pdf (292kB)
This file has been downloaded 664 times
Attachment: Nitrophenol pages Dictionary_of_Applied_Chemistry Thorpe.pdf (698kB)
This file has been downloaded 771 times
There is more very interesting information relevant to what I will say next about my survey of the literature and its implications for these energetic
diazo compounds generally.
A patent of particular interest is US4329503 particularly interesting is example 6. See also the precedent patent US4115652.
Attachment: US4329503 preparation of 2-amino-4-nitrophenol.pdf (720kB)
This file has been downloaded 687 times
Reviewing the circa 1881 work of Benedikt and Hubl which was doubtlessly followed by Von Herz in 1922 without due attribution to the prior art, it is
significant that Von Herz reported that the preferred precursor for DDNR is the mononitroaminoresorcinol and not the dinitroaminoresorcinol which
would be styphnamic acid, derived from the partial reduction of styphnic acid, which is analogous to picramic acid derived from partial reduction of
picric acid.
The interesting aspect about the diazotization method of Benedikt and Hubl is that it is a combined reaction which not only diazotizes the amino
mononitro phenol, but simultaneously adds 1 nitro group, resulting in a diazodinitrophenol as the product instead of the expected
diazo-mono-nitrophenol. This unexpected result could be general as a reaction, so that it is not necessary to partially reduce a trinitro compound
like picric acid or styphnic acid to a dinitro-mono-amino compound and then diazotize.
What is suggested is that ordinary o-DDNP may be possible to make from a mono-nitro-aminophenol derived from a partial reduction of a dinitrophenol by
applying the diazotization method of Benedikt and Hubl which could simultaneously diazotize the 2 position amino while adding a second nitro at the 6
position, or the 4 position, depending on the structure of the 2-amino-(4-nitro or 6-nitro)-phenol precursor used. This general reaction may proceed
without preference for any particular ring position amino to diazotize or any particular ring position to add a nitro, but will generally do both as a
matter of course, simultaneously diazotizing the existing amino and nitrating a vacant or susceptible to nitration ring position in one step. This
would be half confirmed if 4-amino-6-nitroresorcinol is subjected to the same diazotization method and the result is DDNR identical as that produced
from 4-amino-2-nitroresorcinol.
Since the diazotization method also works with the usual dinitroamino precursor, it may also serve as a diazotization method that is general and
applicable to picramic acid or isopicramic acid or styphnamic acid or any of their anaolgues.
The possibility exists also for a 2-diazo position for DDNR, which was the best guess of Von Herz regarding structure.
As I said earlier IMO position 6 for the diazo is excluded, and position 4 seems most likely however a 2-diazo seems also possible. I think a
polarized light rotation study could resolve the structural identification for what is technically possible to be a 4-diazo or a 2-diazo, or could be
a dextro-levo racemic combined isomer. DDNR may not be one structure or the other, but both isomers cocrystallized.
Difficulties observed for attempts to obtain good clean crystallizations for some of these diazo compounds suggests the possibility that mixed isomers
of indefinite proportions could be the explanation.
Here is a screenshot from the Von Herz 1922 patent GB207563.
What is described by Von Herz regarding the potassium salt of DDNR is what I have termed an unequivocal initiating explosive, having an extremely low
critical mass for detonation unconfined. Earlier there was experimentation by Hennig Brand showing that mixture of lead azide with DDNP would
accellerate the DDNP to detonation, avoiding the usual kindling phase of a deflagration runup to transition to detonation. Likewise a mixture with a
different unequivocal initiator like the potassium salt of DDNR should accomplish the same accelleration for DDNP or for p-DDNP with the advantage
that the composite would be a green energetic. The strontium salt of DDNR is also reported interesting in later patents. Strontium is bivalent which
should make possible formation of a basic strontium DDNR salt or for a coprecipitated mixture of the normal and basic salt of strontium if needed for
control of the sensitivity of the mixture.

[Edited on 7/8/2015 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
From the literature I have reviewed there is shown more than one synthetic approach possible for DDNR using resorcinol as a starting material. These
two general synthetic schemes could be summarized as follows.
[1] The most obvious approach is to first make styphnic acid and do a partial reduction to styphnamic acid and then diazotize, following the same
general synthetic scheme as for synthesis of DDNP.
[2] An alternate approach is to make 2,4-dinitroresorcinol and do a partial reduction to the amino-mononitro-resorcinol which is then diazotized
according to the scheme of Benedikt and Hubl, which also adds the needed additional nitro group.
Both of the above schemes may be accomplished by obtaining the dinitrated resorcinol or trinitrated resorcinol precursor for partial reduction,
through a mixed sulfuric and nitric acid nitration that acts upon a first sulfonated resorcinol to produce the nitrated resorcinol desired. Or in the
alternative to a sulfonation of resorcinol followed by di or trinitration, a dinitroso resorcinol may be first formed and then a further nitration may
be performed using nitric acid, by a process optimized to produce a mixture of both the dinitroresorcinol and the trinitroresorcinol, with either the
dinitro or trinitro product predominating according to the conditions used for the conversion of the dinitrosoresorcinol to a nitro resorcinol.
For such nitration schemes there is a then a separation required for the mixed product, to isolate the dinitroresorcinol or the trinitroresorcinol,
which would be then subjected to a partial reduction to the corresponding amino derivative.
However, such a separation and partial reduction performed separately upon the isolated dinitro or trinitro products may not be strictly required, if
the fate of the amino derivative of either the dinitro or trinitro is to be later subjected to the diazotization method of Benedikt and Hubl given the
fact that either partial reduction derivative will be thereby converted to DDNR.
When the desired ultimate product of synthesis is DDNR, there may be no need to separate and isolate the product components mixture of a nitration
which provides a mixture of the dinitro and trinitro resorcinols to be either one subjected to a partial reduction under similar (or identical)
conditions. The mixed product obtained from resorcinol nitration may require no separation and isolation of its mixed components, if the mixture of
dinitroresorcinol and trinitroresorcinol can be subjected simultaneously to partial reduction, and the product mixture of the amino-mononitro
resorcinol along with amino-dinitro resorcinol is to subsequently be diazotized by a method which will convert either or both intermediates to the
same ultimate end product which is DDNR. So it would seem probable that a third scheme for synthesis is possible where resorcinol is the starting
material, and the diazotization method of Benedikt and Hubl is applied. This possible synthetic scheme has not been described per se in the
literature but is a reasonable conclusion which may be drawn by inference from the reactions described by Benedikt and Hubl.
Avoiding losses from separations of intermediates that may not be strictly necessary, would also provide a shortcut eliminating extra synthetic steps
not required for special handling of two substantially equivalent intermediates, which could make such a combined synthetic scheme the most efficient
in terms of yield of DDNR based on resorcinol.
Presently I am still studying the reactions involved in order to propose combined reaction scheme conditions that would operate as I have contemplated
is possible. My first thoughts about this are that the approach would involve first forming the dinitrosoresorcinol and then subjecting that
intermediate to a HNO3 nitration in the cold to produce a predominately dinitroresorcinol product with the greatest impurity byproduct being styphnic
acid, so that resorcinol conversion is most complete to such a mixture, which could be half of each.
With only effort to separate the mixture of nitrated products, and no effort to separate the two products from each other, the mixture of di and
trinitro resorcinol is subjected to a partial reduction. Both products should be separable as a mixture of their salts, and upon decomposition of
those salts using acid, both will form acid soluble forms in solution due to their shared property of amphoterism. The details of the separation of
such a mixture are not fully worked out yet, but I think such manipulation is possible. The acid soluble forms of both the amino derivatives are then
diazotized according to the scheme of Benedikt and Hubl.
That is a very general description of the synthetic approach I contemplate could work, as a simplified method of synthesis for DDNR.
With regard to the alternate route to DDNR from paracetamol it appears likely that a requirement for acetic anhydride may exist to first convert
paracetamol to the acetate and follow by nitration of the resulting diacetyl derivative, as described by Meldola and Klapotke. The simplifying of
that synthetic approach depends upon the easier approach for synthesis of the intermediate 3-nitro acteamiophen by the method described by Mansunto et
al in the Japanese Chemical and Pharmaceutical Bulletin article, which is unconfirmed and suspected incorrect information, likely to be in error and
misidentifying a 3-nitroso acetaminophen as a 3- nitro, this misidentification being suspected due to a discrepancy in reported melting point.
Whether the probable 3-nitroso acetaminophen of Mansunto may be easily nitrated to the bona fide 3-nitro acetaminophen is unknown. Experiments have
not been done to resolve the issue of probable misidentification by Mansunto so the jury is still out on the unknowns involving the Mansunto et al
article.
[Edited on 7/8/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard

Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Something completely different:
Would something along these lines possibly work? 
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I am not certain but I think it will probably not work with the coupling already present between the diazo and the anhydride oxygen, that occurs in
preference to formation of the oxime, which could form in the diazotization. There was a brief discussion about the oxime of phenol in another
thread.
http://www.sciencemadness.org/talk/viewthread.php?tid=12677
There has been a development regarding the Mansunto et al article regarding the alleged 3-nitroacetaminophen which has now been conclusively
identified as a misidentification of what is probably the 3-nitrosoacetaminophen instead.
http://www.sciencemadness.org/talk/viewthread.php?tid=9722&a...
[Edited on 7/11/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
First of all, thanks to Boffis and Philou for the requested hydroxylamine synthesis from nitrite. I must admit that since I was able to aquire some
99% nitromethane, the nitrite/zinc method suddenly seemed like a lot of hassle.
Hydroxylamine synthesis:
105 grams of 96% SA (1 mole) was added to a container with long reflux condensor attached and put on a hotplate with added stirrer bar. While stirring
at 250 rpm, 61 grams of nitromethane (1 mole) was added slowly. Then the heat was turned on and the solution kept at 90-95 deg. C for 24 hours, at
which point the bubbling of carbon monoxide had largely ceased. The flask was allowed to cool to room temperature, after which 250 ml 96% ethanol was
added, instantly precipitating most of the hydroxylamine sulfate as white feathery crystals. After cooling overnight at -20 deg C, total yield was 59
grams of hydroxylamine sulfate, or 72% of theory. there was still some nitromethane in the condensor, and some may have escaped, IMO the most probable
cause of the lower yield than reported.
Unfortunately, there seems no condensation between o-DDNP and hydroxylamine. Adding a pH 6 adjusted solution of hydroxylamine sulfate/NaOH to o-DDNP
produced pronounced fizzing of the suspension as it was allowed to warm up, presumably nitrogen gas, indicating decomposition of the diazo group. The
same happens, though much more slowly at a pH of 3. Maybe the hydroxylamine coupling with the diazo group is a parasitic side reaction that is more
dominant, for which the formed product is unstable and decomposes releasing nitrogen gas. Judging by the colour, the end product in both cases seems
to be ordinary picramic acid again, although it wasn't diazotized again to confirm.
Ah well... VNS chemistry is still interesting to experiment with, so the hydroxylamine sulfate will be put to good use anyway.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Regarding the synthesis of DDNR:
In a recent experiment I heated 3 grams of isopicramic acid with 20 ml 65% HNO3 to about 50 deg. C. for 4 hours. (At lower temperatures there is no
reaction and/or little dissolution of the isopicramic at all.) I was curious whether this way crystalline pDDNP could be formed for people without
access to nitrite, but also whether this could be a possible route to DDNR.
The reaction and colour changes are very interesting. After reaching 50 deg C. most of the isopicramic has gone into solution, forming a deep
orange-red solution. After prolonged heating, the colour seems to change to more of a deep dark red (1-2h), to a deep brownish red (3h) to an almost
transparent light orange-yellow near the end. It is hard to tell, since the soluble isopicramic nitrate is already a very strong orange-red, but the
side of the beaker did seem to show some dark red compound, which would concur with the colour of the trinitroaminophenol, or compound 6, from the
Klapotke article.
When the reaction mix was allowed to cool down it was poured onto crushed ice, precipitating a bright yellow-orangeish crystalline compound. A small
amount was dried at 50 deg C. on the hotplate, it flashes similarly to DDNP. It is drying now, so no yield yet, though it will be very interesting to
react the resulting compound with KHCO3 or PbCO3 in either ethanol or water, as described in one of the patents regarding DDNR salts Rosco posted a
while back.
In the worst case it may be a diazotizing scheme to pDDNP, allowing good control of crystal formation, and usable for those without nitrite. 
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
My guess is that what you have is probably p-DDNP via one of the "old method"
diazotizations using in situ decomposition of HNO3 for diazotization of the isopicramic acid nitrate by the byproduct "red fuming" HNO3 created by a
little heating and some decomposition of the isopicramic acid probably contributiing. It was an old method of making DDNP to pour over picramic acid
boiling hot HNO3, which caused a vigorous fuming reaction and precipitated DDNP. I think it is posted earlier in the thread as an early history
method for DDNP. It is also the method for one of the Dehn DDNP patents. If I recall correctly there is a possible fire hazard for large batches
which they covered with mineral oil to keep the reaction "smothered" under a layer of oil.
Here is the circa 1868 JCS article by Stenhouse and I'll find the Dehn patent also.
Okay I found the Dehn patent US1460708 and it describes use of the ammonium salt and fuming HNO3 gives quantitative yields, and the nitrous acid is
formed via reduction of the HNO3, which seems very interesting.
I think there is possibly another reference to this reaction I'm not certain.
Update, yes, here is the third reference, Matthew Carey Lea published November 1861 regarding DDNP. Attached also is a less yellowed scan of the same
article.
It is interesting though because it provides a possible means of producing a pure product and for controlling crystal size.
Attachment: Pages 150-151 re DDNP Vol 21 (1868) Journal_of_the_Chemical_Society.pdf (144kB)
This file has been downloaded 678 times
Attachment: US1460708 process for DDNP using nascent nitrous acid from HNO3.pdf (319kB)
This file has been downloaded 582 times
Attachment: Am. J. Science and Arts [32] (1861) pg210,211.pdf (909kB)
This file has been downloaded 590 times
Attachment: Pages from American_journal_of_science 32,1861.pdf (268kB)
This file has been downloaded 562 times
[Edited on 7/24/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Quite possibly the product consists mostly of pDDNP instead of the DDNR, although I wonder why in the Klapotke article 100% HNO3 and Ac2O is used,
while they use 65% nitric and heating for the DDNR analogue, any thoughts on this?
I knew about the fuming NA reaction to produce DDNP, tried it once with freshly distilled NA. It is a very vigerous reaction, with lots of NOx
produced and low yields (30-40%) since most of the product is oxidized, yielding the HNO2 in situ. Very interesting find that the use of ammonium
picramate gives a quantitative yield, quite possibly DDNP is diazotized in high yield when ammonium nitrate is added to the heated HNO3, or a small
amount of concentrated ammonia solution. Definitely worth a try, especially when nitrite is unavailable. The finding that even lower concentrations of
nitric are able to diazotize with heating is very interesting as well, since the slow and controled oxidation to yield HNO2 may provide much higher
yields, and the DDNP is completely solubilized in the spent reaction mixture, allowing good crystallization indeed.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Quote: Originally posted by nitro-genes  | | Quite possibly the product consists mostly of pDDNP instead of the DDNR, although I wonder why in the Klapotke article 100% HNO3 and Ac2O is used,
while they use 65% nitric and heating for the DDNR analogue, any thoughts on this? |
I could venture a guess that there was the awkward and unecessarily complicated route to p-DDNP using difficult and expensive reagents done for proof
of structure and to test the earlier researchers report that the 2,6-dinitro substituents would resist further nitration even when the more difficult
path being used did not make use of isopicramic acid.
| Quote: |
I knew about the fuming NA reaction to produce DDNP, tried it once with freshly distilled NA. It is a very vigerous reaction, with lots of NOx
produced and low yields (30-40%) since most of the product is oxidized, yielding the HNO2 in situ. Very interesting find that the use of ammonium
picramate gives a quantitative yield, quite possibly DDNP is diazotized in high yield when ammonium nitrate is added to the heated HNO3, or a small
amount of concentrated ammonia solution. Definitely worth a try, especially when nitrite is unavailable. The finding that even lower concentrations of
nitric are able to diazotize with heating is very interesting as well, since the slow and controled oxidation to yield HNO2 may provide much higher
yields, and the DDNP is completely solubilized in the spent reaction mixture, allowing good crystallization indeed. |
Yes it is a neat trick and has some very interesting aspects. However there is definitely an unnerving aspect about the process which involves the
reaction mixture itself which clearly qualifies as a Sprengel explosive. This for me would be a white knuckles kind of reaction route uncomfortable
to perform manually because of the "ready to go" nature of the reaction mixture, it would bear some thought about using a remote method of
manipulation and viewing. If that mixture detonates it would not be a user friendly experience for anything near.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Most likely you are right as usual, I was hoping that somehow water content would be a contributing factor. Without extensive knowledge of organic
chemistry, its sort of fishing to me...you have no idea what happens underwater, though sometimes you get the thrill of catching a big one.
Regarding the safety of the heated 65% HNO3 and isopicric, perhaps stronger dilutions would also work, increasing safety, perhaps adding an inert
solvent, or large amount of ammonium sulfate or phosphate salt. I think the danger is of performing the reaction is something to keep in minder,
although using excess of NA containing 35% water may be enough to prevent spontaneous reaction.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
There are some additional attachments to the earlier post where I found the third reference for which I was searching earlier.
I think the use of ordinary concentrated d 1.42 HNO3 should work fine and should be safer than use of fuming d 1.5 HNO3.
I think it is likely that once the nitric acid soluble form of the amphoteric precursor "dinitroaminophenol acid salt" is all in solution, that
addition of a reducing agent like ordinary sugar, or ethanol, or perhaps better paraformaldehyde, sprinkled gradually into the nitric acid solution
could generate the nitrous acid in situ and the diazotization should proceed. In the alternative, sodium nitrite or other nitrite, or an organic
nitrite, or even gaseous nitrous oxides from a gas generator of HNO3 warmed with starch or perhaps paraformaldehyde could work. Addition of an
ammonium salt would tend to counteract the diazotization since ammonia like urea tends to be decomposed by nitrous acid due to the thermal and
chemical instability of ammonium nitrite forming as a transient intermediate in decomposition.
We may be zeroing in on a workable method with one of these synthetic schemes, even possibly avoiding the absolute need for a nitrite for the
diazotization. Not too bad if it works okay.
[Edited on 7/24/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Holy shit!!! It may turn out the nitration of isopicramic with 65% nitric acid
at elevated temperatures does produce some DDNR! Some of the yellow orange stuff was carefully neutralized with one mole equivalent of sodium
bicarbonate with overnight stirring, after which a strong solution of KNO3 was added. Unlike pDDNP, a speck of the resulting dark brown stuff
detonates when heated.
Another equally interesting possibility is that a sort of a neutral potassium nitrate/pDDNP double salt has formed. Maybe crystallization from sodium
perchlorate solutions would be interesting to test. Alternatively, the strong KNO3 solution may have precipitated the presumably more energetic
diazonium nitrate of pDDNP. The latter would be easy to test, since it would dissociate when added to water again.
[Edited on 24-7-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Ha! It would be fortuitous if another error has been found in the literature with the ancient reports that the 2,6-dinitro compound the
acetylisopicramic acid or the isopicramic acid itself absolutely resists any further nitration to a trinitro derivative. If that is in fact found to
not be so then yes indeed either DDNR or an isomer of DDNR may be produced. Such a development could indeed be very interesting.
I have wondered if the third nitro group could be introduced by some method different from the extreme methods which were tried, maybe at some
different temperature or reaction time or even by the use of some catalyst that could effect the introduction of the third nitro group. Water content
of the nitration mixture may be that "catalyst" which because it seems counter intuitive was a factor that was overlooked by earlier researchers. It
is entirely possible that a certain water content in the nitration acid mixture will result in a "more aggressive" nitrating effect than is seen with
a lower H2O content nitrating acid mixture. There can also be a catalytic effect for HNO2 acting in concert with HNO3 and some H2O in mixture
particularly at elevated temperature, which results in greater nitrating effect. Such factors could be operative here for this reaction scheme.
Earlier researchers may have never found the niche condition required for the further nitration to occur and wrongly concluded that it is not possible
to further nitrate the 2,6-dinitro-4-aminophenol (isopicramic acid) when in actuality it is possible under different conditions than what they tried
which produced negative results.
It seems possible that perhaps the p-DDNP or the diazonium nitrate salt could form first and that could allow for the third nitro group to enter so
that what may be occurring is essentially a further in situ nitration of p-DDNP or its soluble diazonium nitrate salt precursor. Possibly this is an
exception observed for the synthetic scheme being used which actually isn't a further nitration of isopicramic acid, but a further nitration of the
diazonium derivative of isopicramic acid which must form first as an intermediate for the further nitration to occur. If this does occur it may or
may not be possible to first isolate p-DDNP and simply dissolve it in HNO3 for further nitration. That approach may work or it may not. If not, then
it would point to the soluble diazonium nitrate salt precursor and intermediate to p-DDNP that is being capable of adding the third nitro before that
"upgraded" diazonium nitrate salt is decomposed to the associated diazo-oxide on dilution with water.
Really the probable existence of the soluble diazonium nitrate salt as an intermediate for the p-DDNP has not been described in the literature, but I
have hypothesized that it does probably exist and may be of interest particularly if an insoluble perchlorate salt could be isolated. The diazonium
salt precursor as a soluble transient intermediate has been identified in the literature for similar compounds, as being hydrolytically unstable and
the soluble diazonium intermediate decomposes to and precipitates as the diazo-oxide when the highly acidic diazotization reaction mixture is diluted
with H2O.
If a third nitro is being introduced during the diazotization of soluble isopicramic acid nitrate in a strong nitric acid solution, then the same
effect is being seen as with the method of Benedikt and Hubl where a combined diazotization and nitration addition of one nitro group is occurring
simultaneously.
That third nitro after being introduced is unstable and upon decomposing to a hydroxyl would give DDNR or an isomer, which indeed would be consistent
with your reported observation.
Reportedly the Magnesium salt of DDNR has good solubility which makes it useful for double decomposition reactions to produce the potassium and
strontium and other less soluble salts of DDNR. This may provide an additional physical means of identifying suspected DDNR to maybe do a long
stirring with MgO or Mg carbonate or precipitated Mg(OH)2 to see if the very soluble Mg(DDNR)2 forms. To that solution you can add a solution of the
desired ion like K, or Sr, or Pb, ect. as one of its soluble salts and the lower soluble corresponding DDNR salt should precipitate.
You very well could have found a niche reaction condition that leads to DDNR from isopicramic acid.
Wow! If this is the case what reactions are occurring .......
this is NOT previously published AND is very interesting
This appears to be novel and is very likely a new discovery.
How about that! Well done!
[Edited on 7/25/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Well, after is has dried, yield was only 1.2 grams pDDNP/DDNR from 3 grams of isopicramic (made by AN/SA nitration and deacetylation). Furthermore,
the product after neutralization and precipitation with KNO3 does not behave as unequivocal as the pure potassium salt of DDNR does. Behavour on
heating is different though and detonates even in very small quantities. Could be a mix of pDDNP and/or other products, including DDNR. What colour
does the potassium DDNR salt have, according to the article posted above it should be bright yellow? If the product is a mix, the brown colour may be
from isopicramate impurities due to parital destruction of the diazo group, since in that case excess base was used for prolonged reaction time.
Maybe after dissolution of the isopicramic acid, the reaction mix is better kept at a lower temperature for some time, to keep oxidation to a minimum.
I must say, I doubt if this would be a conveniant route to DDNR, if it formed at all. Under the reaction conditions utilized, a lot of things happen
probably.
I'll try the reaction again soon, using different reaction conditions.
[Edited on 25-7-2015 by nitro-genes]
[Edited on 25-7-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Should be light yellow for the color for DDNR and for the K salt
There is a different brown color noted after recrystallization which is odd.
Klapotke's crew should be burning the midnight oil about now
If you are going to try variations on the experiment, using sodium nitrite or if available potassium nitrite added to the nitric acid solution of
isopicramic acid could accomplish the diazotization at lower temperature and produce a higher yield.
My intuition is that if a third nitro is entering then it is occurring subsequent to the diazotization which is probably favored at lower temperature.
So getting the diazotization done at the lower temperature and then warming to a gentler lower temperature for adding the third nitro may result in
less oxidation losses if that was the reason for a low yield from the longer heating of the reaction mixture.

Attachment: Benedikt and Hubl JCS 1881 Styphnamic Acid and DDNR related pg1133.pdf (264kB)
This file has been downloaded 575 times
Something I just noticed is that the Klapotke article has published now apparently with the errors that were identified in the preview article not
corrected.
In a post I made last month
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
and in earlier and subsequent posts there were errors identified which I expected would be in the preview article that would be reviewed and
corrected, but those errors remain.
I suppose our friends in Munich are not reading us or aren't paying attention in class. 
http://onlinelibrary.wiley.com/doi/10.1002/ejoc.201500465/ab...
[Edited on 7/25/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Interesting read from "organic chemistry of explosives", it seems diazophenols are presumably formed through a primary arylnitramine from nitration of
the corresponding arylamine. Although the reaction is only described for methyl and chloro dinitroaminophenol, would this somehow be possibble for
(iso)picramic acid, or does the hydroxy group lead to excessive oxidation and HNO2 formation? Maybe adding urea or even ammonium nitrate? Ideas anyone?
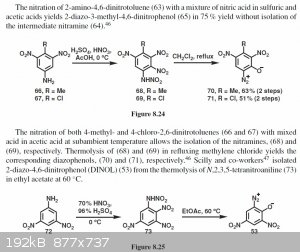
Attachment: organits_chemistry_of_eksplosives__agraval_j._p._hodgson_r._d._-_2007_g._418_s..pdf (4MB)
This file has been downloaded 2894 times
[Edit: Didn't see the book in the SMDB library, so I uploaded it here, if is has already been posted, it can be deleted again, or transfered to the
library]
[Edited on 27-7-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Many diazo compounds are sensitive to decomposition by bases, so it may be better when making the soluble form of suspected DDNR to digest the
material with sodium acetate or magnesium acetate instead of using sodium bicarbonate or carbonate since the buffering effect of the acetate will be
less likely to cause partial decomposition of the diazo compound which could reduce yield. This problem of hydrolytic sensitivity has been reported
for DDNR and use of sodium acetate resolved that issue. I think the DDNR can actually be boiled with sodium acetate without decomposition.
In an earlier post last month a couple of pages back
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
Note that the actual 6 position nitro is shown in error as 5 nitro.
The dense, orange, crystalline sodium salt described is the sodium salt of DDNR, with the 3 hydroxyl obtained by the decomposition of a 3 nitro
residing on the precursor 4-diazo-2,3,6-trinitrophenol, dense, yellow, micro-crystalline powder.
.bmp - 889kB")
There is a theory I have about what may be occurring is indeed a combined reaction that involves diazotization and then subsequent nitration occurring
at ring position 3 or possibly 5, with the third entering nitro being unstable and susceptible to decomposition to a hydroxyl. If the third nitro is
entering at 3 as seems to be likely, then DDNR is the probable result. If the third nitro is entering at 5 then a mirror image isomer of DDNR is
perhaps possible, but I believe it likely that the same DDNR results in either case. If you look at resorcinol as the conventional nomenclature from
antiquity associating it with phenol having the first hydroxyl at 12 o'clock position 1, the second hydroxyl is position 3, meta orientation. But if
you draw this on tracing paper and flip the page over then the reverse perspective shows 1,5 = 1,3 as functionally synonymous expressions.
Here we have with isopicramic acid only 2 possible ring positions which a third entering nitro could occur either 3 or 5, because all other positions
are occupied, and that third nitro on decomposition to hydroxyl will result in the same resorcinol meta oriented pair of hydroxyls where 1,3 = 1,5 and
vice versa.
In my opinion, DDNR is the probable result or most likely product from a combined nitrosation of isopicramic acid with subsequent nitration which adds
a third nitro at 3 which subsequently decomposes to a hydroxyl at 3.
The orienting effect of the diazo at 4 would be ortho-para but the (para) 1 position is already occupied by a hydroxyl so the (ortho) 3 or 5 would be
where an entering nitro would occur as promoted by the diazo, aside from the structural necessity that those are the only possible two vacant
locations, with both leading ultimately to DDNR.
Evidently the diazo at 4 is more promoting of nitro substitution than is the amino group at 4 before diazotization, and the conjugate linkage
subsequently of the diazo at 4 with the hydroxyl at 1 to form the anhydride as a cross-ring linkage may even further promote the entering nitro at 3
or 5 which are essentially synonymous and lead to DDNR after the entering nitro is hydrolyzed to a hydroxyl.
This sequence of reactions would be likely favored by specific pH and temperature and holding times to produce highest yield as is generally true for
many reactions. The optimum temperature for the diazotization may be lower, with a specific warmer temperature and holding time that is optimum for
the nitration and possibly hydrolysis combined. There could actually be three sequential reactions done as a one pot process. Or breaking the
process into stages may work better.
Really I don't mean to harp on this, but I think the case is pretty convincing for the diazo at 4 and the Klapotke article is simply wrong about DDNR
being a 6 diazo which would require some radical and unlikely diazo rearrangement. Before this is done with there is going to be IMO a revision of
structural identification done by Klapotke same as occurred a hundred years ago for Meldola History is going to repeat itself like deja vu ....all over again

The occurrence of diazotization with nitration also occurring subsequently or possibly at the same time has been reported for reactions involving
other subtrates. It seems possible that a nitration catalyst could possibly be useful also for promoting the introduction of an added nitro group,
that the presence of certain salts in the nitrating acid could increase the yield.
In the excerpt below it is mentioned that even nitric oxide can in some cases effect diazotization, which led to my suggestion that paraformaldehyde
added to the strong acid solution of isopicramic acid nitrate may serve to diazotize, since in strong acid that is the result, but I think the nitric
oxide itself then reacts with HNO3 to produce NO2 which likely does the work, similarly as if NaNO2 had been added, which is another option.
References are sparse but according to one I have read Paraformaldehyde with HNO3 reacts differently according to the concentration at 0.5N to 2N
there is NO2 produced directly but at higher concentration NO is the result. See attached article.
Attachment: nitric acid plus formaldehyde.pdf (78kB)
This file has been downloaded 751 times
Here is a screen shot page 10, Chemistry of the Diazo Compounds
https://books.google.com/books?id=XYhY9AUzVD0C&pg=PA10&a...

From the reaction of isopicramic acid heated in nitric acid it would appear there are 3 likely possible products all 3 being energetic.
[1] p-DDNP 4-diazo-2,6-dinitrophenol-(1-anhydride)
[2] 4-diazo-2,3,6-trinitrophenol-(1-anhydride) as a probably transient intermediate having an unstable 3 nitro decomposing to a 3 hydroxyl
[3] DDNR 4-diazo-2,6.-dinitroresorcinol-(1-anhydride)
If what is produced is a mixture which defies easy separation it could still be a useful energetic material as a mixture. But it should likely be
possible to resolve such a mixture using sodium acetate since there is formed a soluble salt of the DDNR. Boiling with magnesium acetate or magnesium
oxide should work as well, perhaps better to form the soluble magnesium salt of DDNR. The other materials, including unreacted isopicramic acid have
extremely low solubility in H2O. According to the Hagel and Redecker patent US4246052 the mixture should be kept slightly acid pH 6.5 and not
excessively neutralized to basic, to avoid decomposition.
[Edited on 7/28/2015 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
possible catalyst for 3-nitro acetaminophen and DDNR
There has been some progress to report a possible usefulness of molybdenum as a nitration catalyst promoting nitration at the 3 position for the first
entering nitro group for a nitration of acetaminophen. Hopefully the same promoting effect occurs likewise for higher nitration attempts on p-DDNP
which has the same promoted substituent location available for a third nitro.
| Quote: Originally posted by PHILOU Zrealone | | Quote: Originally posted by solo |
Requested by Rosco Bodine
Mild, Efficient and Selective Nitration of Anilides, Non-Activated and Moderately Activated Aromatic Compounds with Ammonium Molybdate and
Nitric Acid as a New Nitrating Agent
Sariah Sana, K. C. Rajanna, Mir Moazzam Ali, P. K. Saiprakash
Chemistry Letters
Vol. 29 (2000) No. 1 P 48-49
|
Just to point out that at the end of the document there is a reference nr 12 that is not a reference but a full paragraph of the actual procedure for
the nitration at the wrong place.
Besides this it is very interesting since I have several kilograms of NH4 molybdate and now I know what to do with it
|
Thanks to solo for the file. The file is actually a pdf so here is a corrected format that should save and open as a normal file.
What I tried there changed back to a text file again when I uploaded the file. The encrypted file seems to download as a text file but will open with
Acrobat and can be saved as a pdf and thereafter should open normally.
Attachment: Mild, Efficient and Selective Nitration of Anilides, Non-Activated and Moderately Activated Aromatic Compounds with Ammo (50kB)
This file has been downloaded 650 times
The substituted ring position for the 4-hydroxyacetanilide (synonymous with acetaminophen / paracetamol) would be 3 or 5 (for the hydroxyl 1 as a
phenol) which is a change for the usual first entering nitro absent any catalyst. So in the case of nitration of acetaminophen the first entering
group would occupy 3 or 5 which is the desired meta position with respect to the phenol hydroxyl at 1 and allows for the nitration to proceed easily
to the 2,3,6 trinitroacetaminophen which is exactly what has been desired to be found.
The nomenclature translates what is ortho with respect to the acetamino at 1 is simultaneously meta for the hydroxyl at 4.
The hydroxy acetanilide is an inverted expression for acetaminophenol.
Ordinarily for the nitration of acetaminophen the first entering nitro occurs at position 2, and the second nitro at 6, and then nitration proceeds no
further. However if the first entering nitro can be made to occur at 3, then the second nitro enters at 2, and yet a third nitro enters at 6.
So if the molybdate catalyst steers the first entering nitro to a meta position, with respect to the hydroxyl as it does, then the nitration proceeds
further to ultimately provide a higher nitrated 2,3,6-trinitroacetaminophenol product.
The ultimate product is very likely to be DDNR via the 2,3,6-trinitroacetaminophenol, followed by deacetylation, and subsequent diazotization.
The same ultimate DDNR product may be more easily obtained if the same catalytic effect of molybdenum applies as is anticipated to promote a further
nitration of p-DDNP resulting ultimately also in DDNR.
It is interesting how a molybdenum salt could operate as a nitration catalyst. There have been reported other possibly useful aromatic nitration
catalysts to include tungsten salts I'm certain, and some other metal salts which may be useful also mentioned in a patent for which I'll search. I
know this could possibly be useful with regards to increasing the level of nitration of already partially nitrated aromatics which generally resist
further nitration and help get the job done under milder conditions for the nitration.
Here it is GB501034
Attachment: GB501034 nitration catalysts.pdf (212kB)
This file has been downloaded 560 times
[Edited on 8/6/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Seems old Rosco was right as usual regarding the solubility of the isopicramic acid sulfate after deactylation. The deacetylation mix just needs stronger dilution to precipitate all of the dark
brown/black impurities. When the deacylation is performed in more strongly diluted sulfuric acid and is allowed to cool down completely before
filtereing, all of the isopicramic acid stays in solution and a much purer product is obtained when titrated with a base to obtain the free and
insoluble isopicramic acid again. The resulting isopicramic acid is very pure and gives fine glittering needles of p-DDNP when diazotized using hCl
and sodium nitrite.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Version 2.0:
Synthesis of 4-amino-2,6-dinitrophenol (iso-picramic acid) from acetaminophen
Notes: Look for the cheapest brand of Tylenol/Paracetamol tablets, containing the highest amount of acetaminophen (usually 500 mg). Weigh 10
tablets or so to determine the percentage of fillers present and compare different brands for the lowest percentage. Additionally, this extraction
procedure may not work equally well with every brand of tablets and would depend on the composition of the binders/fillers. If the nitration produces
some sticky residues there are likely remaining impurities present from the tablets. Alternatively, pure acetaminophen is also available commercially,
which would eliminate the extraction procedure.
Extraction of acetaminophen from Tylenol or Paracetamol tablets
Collect a number of tablets, translating to a combined weight of 25 grams of pure acetaminophen, (so weight of fillers not included) and grind as fine
as possible using a coffee grinder. Transfer the powder to a 250 ml beaker and add 100 ml of denatured spirits. Heat the mix under reflux to the
boiling point on a hotplate or boiling water bath and let it stir for about 5 minutes with gentle boil to extract the acetaminophen. Let the solution
cool to room temperature and filter the solution into a 250-500 ml beaker, which will remove most of the fillers.
Next, put the beaker on a hotplate and put it on the highest temperature setting and let the solution boil while stirring. The ethanol fumes are very
flammable and potentially explosive so do this outside! When the solution has evaporated to about half of its original volume, add 50 ml of water
while keeping at boil. When nearly all of the ethanol is evaporated (takes about 15 minutes) the acetaminophen will precipitate over the course of
10-15 minutes as a course sand like precipitate. Take the solution from the hotplate and leave it to cool to room temperature. Add another 50 ml of
cold water and filter the suspension. Wash 3-4 times with cold water and filter to collect the acetaminophen. The filtrate will have a slight yellow
tint and appears milky, this is normal. Let the acetaminophen dry at room temperature for 24 hours, to obtain pure crystalline acetaminophen as a free
flowing powder. (21-22 grams)
Nitration to dinitro acetaminophenol
Prepare a large cooling bath containing crushed ice and water, containing at least 500 grams of crushed ice. Put a 100 ml beaker on a scale and poor
in 70 grams of 96-98% sulfuric acid. Transfer to the ice bath and let it cool to 0-10 deg. C. Then add 25 grams of dry ammonium nitrate in small
portions, keeping the temperature below 20 deg C. Swirl or stir for about 5 minutes to dissolve all the ammonium nitrate and keep on ice.
To a separate 500 ml beaker, add 100 grams of conc. sulfuric acid and also transfer to the ice bath, let it cool to 0 deg. C. Add a stirrer bar and
thermometer, and put the ice bath on a stirrer plate, set at 125-250 rpm. Weigh out 20 grams of finely powdered acetaminophen and add small portions
at a time to the sulfuric acid, while keeping temperature below 10 deg C. Let it stir for an additional 10 minutes after the last addition until
everything has dissolved.
After the acetaminophen/SA solution has reached 5 deg C, slowly start adding the ammonium nitrate/SA solution. It is best to keep the temperature
around 5 deg C during the additions. The nitration responds very rapidly upon the additions, about 2-3 ml with each addition is about the maximum that
can be added for the first additions, resulting in a jump from 5 to 10 deg C. Above 15 deg C, foaming starts to become evident, probably due to
decomposition of the acetaminophen.
Depending on the efficiency of the cooling bath, the total addition will take about 30 minutes. Halfway the nitration, the solution will attain a dark
brown colour with each addition, which will fade to a more orange colour again after some time. After the final addition is made, let the solution
stir for another hour while keeping on ice. Finally, ad 250 grams of finely crushed ice (preferably directly from the -20) to the nitration mix. Some
foaming will occur, let the solution stir for another 20 minutes while in the ice bath until the foam has mostly dissipated. The dinitro
acetaminophenol will settle as an yellow-orange precipitate to the bottom of the beaker, it is filtered and washed with ice cold water from the ice
bath. Wear gloves while handling it, as it is a very strong dye! Under acidic conditions it gives a bright yellow colour, while at near neutral pH a
very strong grapefruit like colour is obtained to a deep red/violet when further basified.
Note: Yield can be increased by neutralizing the filtrate with concentrated ammonia solution and leaving overnight in the fridge, precipitating
the residual dinitro acetaminophenol as the matt red coloured ammonium salt.
Deacetylation to isopicramic acid
After filtering and washing, the dinitro acetaminophenol can be directly transferred to a 500 ml beaker. Set stirring to a low setting of about 100
rpm and add 137.5 ml water + 12.5 ml of concentrated sulfuric acid. Heat the suspension to 95 deg C. for about 1 hour under reflux. Monitor the
deactylation and use a pipet to flush down remaining dinitro acetaminophenol from the side of the beaker. The yellow-orange suspension will gradually
go from orange to a deep dark red over time and the initial suspension will form a solution of the soluble iso-picramic acid sulfate. Take the beaker
from the hot plate and allow to cool down to room temperature. Filter out any dark coloured impurites, and transfer the clear filtrate into a 500 ml
beaker, and put it on the stirrer again. Set stirring to 500 rpm, and add small portions of household ammonia over the course of 15-30 minutes, until
the solution is just slightly acidic at a pH of around 4. The iso-picramic acid will separate as 18-21 grams of a brick red glistening crystalline
precipitate that should filter very easily from gravity alone. After drying the product should be an orange-brownish in colour. Over neutralization
from adding too much ammonia will give a more brownish colour due to the presence of the purple coloured ammonium isopicramate, which can affect both
storage stability and diazotization, thus is best avoided.
Notes:
1. Filtering of the deacetylation mix after cooling down is very important to remove insoluble impurities from remaining tablet-binders or
by-products from the nitration itself, otherwise a clean product will not be obtained.
2. The isopicramic acid can be recrystallized from boiling ethanol (solubility about 3 grams per 100 ml) by slow addition of ice cold water and/or
boiling down, upon which the isopicramic acid will precipitate as fine plate like crystals having a beautiful golden appearance.
3. Alternatively, the isopicramic acid/ sulfuric acid solution after deacetylation and filtering can be directly used for the diazotization using
sodium nitrite at 0 deg C. giving fine orange-brownish needles of p-DDNP.
[Edited on 9-8-2015 by nitro-genes]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Very nice, glad you got the issues with the deacetylation and filtration sorted out.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Thank you for the detailed report. This is one for the books, and provides valuable information not found anywhere else.
Regarding the purification of the isopicramic acid by recrystallization, I think the early literature reported also that recrystallization from HCl
worked very well via the formation of a soluble isopicramic acid hydrochloride, which left impurities undissolved. Then upon near neutralization of
the filtered acidic solution the pure low solubility isopicramic acid precipitated.
It would be interesting to see if isopicramic acid or p-DDNP would form a dinitramide or a dinitramide double salt. A similar idea was brought up
earlier about a possible perchlorate which could also be interesting.
Here is US5976483 for the nitration of sulfamic acid commonly available as tile cleaner to produce dinitramide or dinitramidic acid, HN(NO2)2 which
could also have other interesting uses.
Definitely a project for a cold arctic winter with the low temperature requirement.
Actually the sulfamic acid precursor can be used as a getter and decomposer of nitrous acid in a nitration mixture similar to but more efficient than
urea, so the sulfamic acid could be useful for other things also, and it might result in a different result entirely for some nitrations possibly
including the nitration leading to acetylisopicramic acid. This is a complete unknown as to what may result and no search has been made, so it is
just an idea I thought could be useful was worth mentioning. I found a patent US5955050 describing this use of sulfamic acid as an NO2 scrubber which
says that it only works in aqueous H2SO4 of lower than 70% strength. So it wouldn't work as an NO2 inhibitor / antioxidant in concentrated H2SO4 for a
low H2O content nitration mixture, based on that information.
Attachment: US5976483 Dinitramides from Sulfamic Acid Nitration.pdf (744kB)
This file has been downloaded 597 times
[Edited on 8/10/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks, after having performed the nitration of acetaminophen again to update the synthesis protocol posted above, there are some interesting things I
noticed about the nitration and the formed DNAc. It occured strange to me that the DNAc direcly after filtering from the spent nitration mix has a
definite orange colour to it. When a small amount of the water diluted nitration mix was allowed to stand at 4 deg C. overnight, the colour had
changed to a more golden yellow colour, which corresponds better with the earlier reported "straw yellow" colour of the DNAc from literature.
I always assumed that this may be an effect of crystal shape, since the orange colour is nearly absent when using HNO3 instead of AN. It occured to me
that a lack of water and oxidation of some of the acetaminophen may actually produce quite some HNO2 in situ, which could produce N-nitroso DNAc (or
another compound?). To test this possibility, a small amount of the straw coloured DNAc was acidified (no colour change), after which NaNO2 solution
was added, immediately producing the bright orange colour again. The colour is almost identical to to the presumed 3-nitro(so) acetaminophen described
in the Japanese paper. Since the latter is immediately destroyed when attempting deacetylation in hot conc. SA, leading to fizzing of the solution and
ultimately only resulting in brown crud. The dinitro N-nitroso DNAc (or other product from HNO2 reaction), is likely the main impurity, that upon
deacetylation produces the dark brown impurities. What would be the likely decomposition products of the N-nitroso destruction in SA, presumably, it's
decomposition would be similar to other secondary N-nitroso or nitramine compounds? Would a higher water content in the nitration mix possibly lead to less of the N-nitroso product?
Some things I thought of:
1. Caution is advised when handling the synthesized DNAc, since N-nitroso compounds are usually carcinogenic
2. The nitration using AN instead of NA may need even lower temperatures to prevent oxidation, or perhaps adding urea (or performing the nitration
with urea nitrate/nitrourea) would help
3. Apparently, upon standing in diluted acid the N-nitroso DNAc decomposes/hydrolyses to produce DNAc again, is this normal behaviour for a
N-nitroso? (trying to confirm the N-nitroso is the most likely impurity)
4. Although Meldola has shown that DNAc cannot be nitrated further, it seems strange that DNAc when added to conc. nitric acid/Ac2O doesn't form the
secondary N-nitramine, which could rearrange to a 3-nitro derivative. Is the the result of the keto-enol tautomerism of the OH group?
5. Can the N-nitroso DNAc alternatively be oxidized with H2O2?
[Edited on 11-8-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Great minds think alike. The possibility of improving the yield by reducing
any nitrous acid present in the nitrating acid was exactly what I was thinking about when looking at sulfamic acid as a possible addition of 1-2% to
the nitration mixture, and urea was another possibility, and possibly yes H2O2 may even be best of all for a low temperature. It could be an
advantage to change the order of addition and add the acetaminophen in H2SO4 to the nitrating mixture. That would be the first thing to try to see if
that will provide any improvement. The thing being nitrated can have a reducing effect on the nitric acid and that effect could be greater when the
ratio of the HNO3 is low, as it would be when the HNO3 is being added gradually to a mass of the material to be nitrated, as opposed to the reverse
order of addition where the HNO3 would be in excess. It can in some cases change entirely what is the result of the reaction simply to change the
order of addition. Avoiding intense sunlight is a good idea too because there can be photosensitivity for such mixtures as is true for HNO3.
With regards to the higher water content actually being of benefit in a nitration mixture by opposing the formation of nitrosylsulfuric acid, yes it
is possible to occur that way. Some trinitrations can occur in systems that are 90% H2O but it requires near boiling temperatures and not too easily
decomposed materials being nitrated. However it is unknown if the nitration mixture would withstand dilution to 30% H2O where the NO2 nullifying
effect of sulfamic acid may be observed, and what would be the effect on temperature requirement for a warmer range or holding time is also unknown
and the changes could be counterproductive.
Generally higher H2SO4 content and lower H2O content allow for a lower temperature for nitration and DNAc may not withstand a higher temperature
nitration, so it could just end up being a tradeoff what is the optimum nitration mixture and temperature and holding times. Nitration schemes are
nuanced by the composition and time and temperature that makes some interesting process algebra to find what is optimum.
[Edited on 8/11/2015 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
What you could be observing with the color shift is a dye indicator pH effect similar as occurs with picric acid at extreme low pH the color fades
even completely and then at an increasing pH yellow appears which becomes more orange colored as the pH increases still more even for a solution that
is still extremely acidic, the transitions occur at a very low pH. When you add NaNO2 there is a distinct alkalinity about NaNO2 which shifts the pH
higher and the orange color appears, and then when you add acid, even HCl should work, the color fades again if a pH indicator dye effect is what you
are observing. The color density of yellow dye appears red-orange to rust brown color in concentrated form and will appear yellow on dilution or if
the crystals are crushed and smeared out in thin layer. Red crystals of potassium ferricyanide show the effect of color density "red shift" as does
normal lead styphnate, red brown rust color crystals and a yellow to yellow orange solution.
It could be that the nitration at low temperature is not going to completion and the ratio of nitrating agent in excess of theory may need tweaking or
the holding time and temperature or both may be changed slightly to minimize any byproduct formation. It can take many repeated experiments carefully
charted to tweak a nitration to optimize it for producing the most pure product and it will change things when a nitrate salt plus sulfuric is used as
compared with neat HNO3 plus sulfuric.
|
|
|
| Pages:
1
..
18
19
20
21
22
..
33 |
|








