| Pages:
1
2
3
..
33 |
Madog
Hazard to Others

Posts: 221
Registered: 20-5-2002
Location: USA
Member Is Offline
Mood: lysergic
|
|
DDNP & related compounds: The über thread!
DDNP seems like a perfect primary, relitively easy to make for the home chemist and powerful, not too sensitive and most importantly storeable. i
tried to maek it myself:
i made around 9g of picramic acid via the method shown in books that you find about primarys( Improvised Primary explosives, KIBC, etc) and then i
used the ratios on megas site to make the HNO2 solution, to this i added 5g of picramic acid. the reaction was quick with stirring, and lots of
yellow/light brown precipitate was formed, this was filtered and dryed, it burns like described but i tried to confine some with no luck. i tried the
procedure again only i disolved 4g of the picramic acid in around 250ml of water, this seems very strange that it disolved, picramic acid is only
0.1g/100ml but sodium picramate is 2g/100ml. i got the same precipitate as last time, acts the same too.
i got about 3 grams both times, i wonder what it is, will explode from shock but takes a nice blow! perhaps i should try confineing it more, i didnt
think DDNP would need so much confinement, it is also soluble in water, DDNP is not. the PH of a solution of it was about 6, but im thinking its
actualy neutral considering it was made with acid and unwashed.
anyone have any idea what it is? perhaps the "picramic acid" was sodium picramate? the picramic acid was nice red and burned nicely leaveing lots of
ash, it didnt burn with a soft flame. the product burns the same only alot faster and leaves less ash.
any ideas?
[Edited on 17-6-2015 by Bert]
Most people outgrow their pyro tendencies, we are the ones who\'s tendencies outgrew us.
|
|
|
Nick F
Hazard to Others
Posts: 439
Registered: 7-9-2002
Member Is Offline
Mood: No Mood
|
|
"relitively easy to make for the home chemist"
Haha, [ironic] lol.
Sodium picramate is red, picramic acid is more brown.
You may have a sodium picramate/picrate mixture I guess, probably with a bit of DDNP and other crap. It sounds like the product I normally get, which
is not DDNP I think.
The few times that I've had DDNP form, it often forms in small dark reddish brown grains consisting of masses of microscopic crystals, resulting in
round grains about 0.5mm accross, which are very convenient as they are free-flowing. I don't know if this is normal.
It burns almost as fast as guncotton, producing a little black mushroom cloud of soot, that hangs in the air like when you burn acetylene without
extra oxygen. It also produces radiating soot stains on whatever you set it off on.
|
|
|
Madog
Hazard to Others
Posts: 221
Registered: 20-5-2002
Location: USA
Member Is Offline
Mood: lysergic
|
|
ok, hmm, well, it seems easy. next time i make picramic acid im gona try treating it with HCl before filtering. then washing with cold water. picramic
acid shouldnt disolve like that. i think your right about my product at least to some extent, it leaves yellow stains like TNP does so im thinking
some is TNP or Na picrate. the Na picrate was disolved in manufacture of picramic acid(for the most part) but i did and still do have a couple little
rocks of Na picrate in the product.
Most people outgrow their pyro tendencies, we are the ones who\'s tendencies outgrew us.
|
|
|
andreas
Harmless
Posts: 7
Registered: 19-8-2002
Location: Overijsel
Member Is Offline
Mood: No Mood
|
|
Has anybody noticed a so2 smeel when burning their endproduct.
It's obiously not ddnp.
As soon as I make my nitrate addition machine I'll make up some tnp to make some ddnp. I have received some commercial nitrite, but is their a other
way to reduce the picric acid to picramic.
|
|
|
Madog
Hazard to Others
Posts: 221
Registered: 20-5-2002
Location: USA
Member Is Offline
Mood: lysergic
|
|
i have also noticed the SO2 smell
Most people outgrow their pyro tendencies, we are the ones who\'s tendencies outgrew us.
|
|
|
Madog
Hazard to Others
Posts: 221
Registered: 20-5-2002
Location: USA
Member Is Offline
Mood: lysergic
|
|
today i set off the stuff i had, i just put it in a pill bottle with a fuse and chucked it in the backyard, made a nice fireballish flash with a
report, but that stuff as said before is definately not DDNP, it seemed somewhat as senstive as picric acid to hammer
Most people outgrow their pyro tendencies, we are the ones who\'s tendencies outgrew us.
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
DDNP:
Red yellow amorphous powder
Density: 1,63g/ccm
Deflagration point: 180°C
Impact sensitivity: 1,5 Nm (150g from 1 meter or 1,5 kg from 10 cm)!
Sparingly soluble in water!
Soluble in methanol, ethanol!
Very soluble in aceton, nitroglycerin, nitrobenzene, aniline, pyridine and acetic acid!
Darkens fast in sunlight!
Made from picramic acid by diazotation in excess HCl with NaNO2 and efficient cooling!
The dark brown product is then purified by dissolution in hot acetone and reprecipitation with cold water!
Priming abilities:
More powerful than Hg fulminate and a little less than Pb azide!
Some personnal coments:
 
1°)To make picramic acid you need Trinitrophenol; but in TNP synthesis if not strong enought acids are used; you end up with DNP that is also
sensitive HE (less sensitive and powerfull than TNP); it also makes sensitive unsoluble explosives heavy metal salts). DNP and TNP looks pretty much
the same thus!So if you had DNP; you would make a very poor picramic like compound!
2°)When picramic acid is made you may have reduction of ortho, but also of para nitro from TNP...if no purification is made, you have a mix of
picramic acid and of paraamino dinitrophenol that would lead to a diazonium salt of different properties than DDNP!
3°)If you work with an excess of dilluted HCl; the only salt of sodium you will find is NaCl; sodium picramate can't exist in strong acid
solution!
4°)If you don't cool enought the reaction media; you will have oxydation and loss of N2 from the diazo compound and you come back to a
dinitrophenol or dinitroorthoresorcinol!
(O2N)2C6H2(OH)(N2Cl) -H2O/heat-> (O2N)2C6H2(OH)2 + (O2N)2C6H3-OH + N2 + HCl
5°)If you don't recristallise the crude impure product from aceton, you will have poor HE properties!
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
| Quote: |
From: Anonymous
To: (my e-mail address)
Subject: DDNP and organic nitrites
For those of you who are doing experiments investigating diazodinitrophenol DDNP a different approach to the nitrosation may produce a more desirable
product.
The disadvantage of the DDNP end product normally produced from the nitrosation of picramic acid in aqueous solution is that the DDNP precipitates as
a low density amorphous powder unless crystallization modifiers and special recrystallization techniques are employed to improve the density of the
end product.
It has occured to me that a better crystalline form of DDNP might result directly from the nitrosation if the nitrosation of picramic acid or sodium
picramate was performed in alcohol solution and the nitrosation agent is an organic nitrite. In another communication I made reference to ethylene
glycol dinitrite. Because of the ease of synthesis and the high boiling point the ethylene glycol dinitrite (b.p. 98 degrees centigrade) is of
particular interest as a nitrosation agent. See US2166698. I would avoid the use of isopropyl alcohol as a candidate solvent because DDNP reacts with
it. A lower alcohol like methanol may be okay.
After the nitrosation is completed the alcohol could be evaporated or a slow dropwise addition of water could be used to produce a slow precipitation
of the DDNP which should have a better crystalline form and higher density by either method.
I have not attempted this approach for DDNP so this only a proposed experiment which seems logical. There is not enough time for me to pursue
experiments of my own regarding every idea that occurs to me. So I simply pass along these ideas and welcome any feedback from anyone who may do the
experiment.
A s an afterthought related to the gemeral topic of ntrosation organic nitrites may be useful also in producing better yields of R-salt in nonaqueous
sytems and R-salt itself may find use a nitrosation agent in some reactions. |
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
DDNP patents
| Quote: |
From: Anonymous
To: (my e-mail address)
Subject: DDNP related patents
The extent of my own experiments with DDNP revealed that the amount time and of work required can be applied to produce other different initiators
which are superior to DDNP. For any others pursuing further experiments, I can share my list of patents related to DDNP which I have saved from when
I was experimenting with DDNP.
US2396152 and GB568109 describes coprecipitation of nitrated polyhydric alcohols/DDNP useful as flame sensitive detonating compositions.
This process would be unaffected by the crystalline form in which the DDNP was obtained from synthesis, since the DDNP is redissolved for producing
the coprecipitate. I did not do experiments to confirm the findings described by the patent. These particular patents are listed first because in my
estimation they describe the application where DDNP would potentially find any practical usefulness, basically being used as a cookoff enhancer to
decrease the critical mass of sensitive secondaries which are known to undergo deflagration to detonation transition in slightly larger quantity all
by themselves when ignited in confinement.
The conclusion at which I arrived after many experiments with DDNP is that it is one of those materials which is simply over rated in most of the
literature, and easily surpassed by alternate choices in real world performance tests. DDNP is one of those materials which seems to be a darling of
technical writers, but simply fails to make as good a showing in actual tests. If one reads all the related patents and follows all the shortcomings
of DDNP which are attempted to be addressed by various improvements, the story will become clear that DDNP is really not that great. Otherwise there
wouldnt be so many patents which describe ways of trying to improve its density and so forth.
Anyway, the following additional patents are DDNP related:
US1404687 original patent
US1428011 describes DDNP/picric acid eutectic and acknowledges the
decomposition of DDNP by certain solvents.
US1472791 ammonium picramate from picric acid
US1460708 described process seems inherently dangerous
US1952591 DDNP classic synthesis described
US2408059 crystallization modifiers
GB836410 starch modified granulation
GB849101 methylcellulose granulation improver
US2214721 DDNP/chlorate enhanced mixture
US2104513 DDNP/lead nitrate density improved mixture
GB333534 DDNP/lead azide mixture Hehehe Why bother ?
GB333539 DDNP/oxidizer mixtures Not hot enough by itself ?
GB406228 lead dinitrophenylazide from DDNP/ sodium azide/lead nitrate
GB412460 same as above, tried these and they are much inferior to
azo-clathrates so why bother ?
My own experiments with DDNP ended with the conclusion that DDNP basically sucks. And when you need to use a nitrite and picric acid, it is simply a
waste of precursors when compared to using the nitrite(with hydrazine) to get to sodium azide, and then the picric acid being used in an azo-clathrate
synthesis. With regards to DDNP, been there, done that already. Maybe I missed something in my experiments that others will find. But based upon what
I found, I would never recommend DDNP as a worthwhile synthesis. |
|
|
|
Taaie-Neuskoek
Hazard to Others
Posts: 222
Registered: 14-5-2004
Location: Zermany
Member Is Offline
Mood: Botanical!
|
|
Can picramic acid (1-hydroxy 2-amino 4,6 dinitro benzene) not be made by starting from 2-nitrophenol, reduce the nitrogroup to an amino group and
nitrate the 1-hydroxy 2-amino further to picramic acid?
2-nitrophenol is not probably not very easy to make, as the nitrogroup will most likely be introduced at the 4-postion, so separation is needed one or
the other way, at a certain stage.
Under which circumstances would the nitration take place, probably boiling in H2SO4 and HNO3 for a while??
Never argue with idiots, they drag you down their level and beat you with experience.
|
|
|
Sickman
Hazard to Self
Posts: 98
Registered: 9-5-2004
Member Is Offline
Mood: Icy and I see!
|
|
Rosco,
I know you have mentioned in this thread that "DDNP basically sucks" and I would agree with that in the sense that DDNP alone without any measure to
make it more "unequivocal" as you would put it, doesn't make a reliable initiator.
However in my reading of several patents on DDNP it has occurred to me that coating the crystals with dextrin for example, creates confinement of the
crystals which causes them to be more "unequivocal'. However just like lead azide, dextrinating the material doesn't lead to a superior initiator, as
compared to the use of the azo-clathrate, caged lead azide, in US3431156 and your own personal improvement to the patent. Thanks for that by the
way!
Now bear with me here for a moment while I present my idea! In keeping with the thinking that lead picrate is able to entrap a significant amount of
lead azide, I was wondering if lead picrate would not be able to entrap diazodinitrophenol within it's crystal structure in much the same way as it
does with lead azide? The goal of course being to make the DDNP entrapped within the crystal cage of lead picrate more "unequivocal", being detonated
directly from flame as opposed to doing it's typical deflagration to detonation transition, which in the case of DDNP, won't occur without
confinement. My thought here is that the DDNP entrapped within the lead picrate would be confined by the lead picrate, causing an unequivocal
effect.
Now the technical considerations! In my estimation it's seems theoretically possible for the lead picrate or lead picramate, which ever the case may
be, to entrap within it's crystal structure DDNP. However, now I'm trying to come up with a simple method for introducing the DDNP into the lead
picrate structure. At the same time it seems to me that I cannot assume that the lead picrate will respond the same way to DDNP as it does to the
azide.
One possible senario that doesn't really click very well by my reasoning, but may be worth a try, is to go for entrapping the DDNP in lead picramate
as opposed to lead picrate. The method I was thinking was to first prepare the lead picramate by adding picramic acid to an acoholic sodium hydroxide
solution and then adding lead nitrate to this. Then bubbling nitrous oxide through the mixture. However I see potential flaws in this approach, becase
it expects that the nitrous oxide will convert a large portion of the lead picramate to DDNP and simultaneously entrapping the DDNP, into a forming
clathrate of DDNP, with the unreacted lead picramate.
Any suggestion for the entrapment of DDNP in a true clathrate with lead picrate or lead picramate or some other energetic material that like those
afformentioned have the ability to self deflagrate(for sure) and also the ability to be a crystal host to DDNP (maybe) , would be most
appreciated!
Sickman
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
If it had any chance of working it would have to be run in a solvent other than plain water , something like a moist acetone or alcohol or toluene
......and the DDNP would
probably have to added already formed into the hot solution .....no isopropanol because that reacts with DDNP . But I am not sure this would even
work since
the basic lead picrate clathrate structure is based on
a commonality of all components being lead salts which
have a strong tendency to also form double salts .
I am not sure that a non-lead compound would have much hope of merging into that matrix , since it would be
a dissimilar material .
DDNP has a hydrazine derivative and salt that could be more interesting . And the disclosure of methods for slow precipitation and crystallization
of DDNP in denser
form advanced its potential beyond that of the low density material on which I based my observations that
it wasn't very impressive . I think DDNP is probably
useful under very specific qualified conditions where it
is optimized ....but there are easier things to get right
so that they perform predictably well .
Lately I have been looking at some of the polysulfide
patent literature related to production of picramic acid
in order to refine synthesis of that precursor . Pinnning
down good details on that should greatly facilitate any
further experiments with DDNP by making it easy to produce in good quality crystals in good yield .
I think it warrants further experiments but the usefulness
in a clathrate doesn't seem as promising as some of the other possibilities . The clathrates usefulness is really
pretty specific as a means of enhancing the chemical stability of lead azide . But it is the exceptional power
and detonability of lead azide even in that "diluted"
form where it is bound up in a clathrate which lends
the not greatly diminished small quantity detonating
property to the clathrate . DDNP isn't hot enough and sassy enough in small amounts to do for the clathrate
what lead azide does . That's my opinion on it anyway .
|
|
|
braden00
Harmless
Posts: 2
Registered: 10-5-2011
Member Is Offline
Mood: No Mood
|
|
Ok I cut apart a bunch of #11 percussion caps which are used in guns...ive tried using google but i cant find out the compound in them. I think its
ddnp because that seems like the obvious choice as they are non-toxic which rules out lead and mercury compounds. The powder in them is a dark
brown/greenish powder and is pretty sensitive to a hammer blow. Its also seems to be very powerful any idea as to whats inside?
|
|
|
quicksilver
International Hazard
Posts: 1820
Registered: 7-9-2005
Location: Inches from the keyboard....
Member Is Offline
Mood: ~-=SWINGS=-~
|
|
Quote: Originally posted by braden00  | | Ok I cut apart a bunch of #11 percussion caps which are used in guns...ive tried using google but i cant find out the compound in them. I think its
ddnp because that seems like the obvious choice as they are non-toxic which rules out lead and mercury compounds. The powder in them is a dark
brown/greenish powder and is pretty sensitive to a hammer blow. Its also seems to be very powerful any idea as to whats inside?
|
The commercial brand name would have to be researched if a lab was not available to utilize a variety of tests. Most often the formula is a trade
secret and NOT publicly available. "non-toxic" is a very unusual descriptor. I don't believe I can think of any explosive material that is
"non-toxic". But the possibility of it being DDNP due to it's color is low. Color really doesn't mean that much with many commercial products as their
may be another reason such as binder - for it's color, etc, etc.
It could be tetrazene and tetrazene-combinations. There really is no way to know outside of testing the material.
|
|
|
The WiZard is In
International Hazard
Posts: 1617
Registered: 3-4-2010
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by braden00 | | Ok I cut apart a bunch of #11 percussion caps which are used in guns...ive tried using google but i cant find out the compound in them. I think its
ddnp because that seems like the obvious choice as they are non-toxic which rules out lead and mercury compounds. The powder in them is a dark
brown/greenish powder and is pretty sensitive to a hammer blow. Its also seems to be very powerful any idea as to whats inside?
|
You checked for an MSDS?
|
|
|
braden00
Harmless
Posts: 2
Registered: 10-5-2011
Member Is Offline
Mood: No Mood
|
|
I haven't checked but I did test solubility and it was insoluble in water but soluble in acetone which is in line with the behavior of DDNP. also its
friction sensitivity is pretty high once I put it in water and filtered the impurities...I guess theres no way to know for sure just an interesting
though
|
|
|
The WiZard is In
International Hazard
Posts: 1617
Registered: 3-4-2010
Member Is Offline
Mood: No Mood
|
|
Percussion cap MSDS
http://www.chemcas.com/msds112/cas/3955/15245-44-0_109-27-3_...
Blount Inc.
http://glarp.atk.com/2008/msds/Musket_Caps.pdf
CCI
CCI only claims that their #11's use an — Modern non-corrosive, non-mercuric priming mix
http://www.cci-ammunition.com/products/primers/primers.aspx?...
Remington MSDS
http://www.spreadia.com/Percussion_caps/133494963/Remington_...
-----------
&c., &c. Google is your friend.
www.justfuckinggoogleit.com
|
|
|
The WiZard is In
International Hazard
Posts: 1617
Registered: 3-4-2010
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Madog | | DDNP seems like a perfect primary, relitively easy to make for the home chemist and powerful, not too sensitive and most importantly storeable. i
tried to maek it myself: |
Good old Köhler and Meyer 4ed. of which I own a hard copy
(I have an PDF of a latter ed.) cites —
Title: Diazophenols - Their Structure and Explosive Properties
Personal Author: Lowe-Ma, Charlotte K Nissan, Robin A Wilson, William S
Corporate Author: NAVAL WEAPONS CENTER CHINA LAKE CA
Source Code: 403019
Page Count: 33 page(s)
AD Number: ADA197439
A free DL from the usual source.
By da. K&M claim that D.D.N.P. which is a red amorphous
powder is It is of interest for --> Lead-free priiming
Compositions.
Humm a red powder in a Green comp. Chemistry
is funny that way sometimes.
[Edited on 13-5-2011 by The WiZard is In]
|
|
|
KemiRockarFett
Hazard to Self
Posts: 84
Registered: 23-7-2004
Member Is Offline
Mood: No Mood
|
|
Just want to add something about DDNP. I tried the synth long time ago and can say it was not easy.
The terrorist in Norway claimed that he used DDNP as primary in his bombs. Its written in his manifesto. He also writes that diesel fuel is an oxidant
 How many here belive that a man with no chemistry knowledge manage to
synthesise this primary explosive? How many here belive that a man with no chemistry knowledge manage to
synthesise this primary explosive?
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Taaie-Neuskoek | Can picramic acid (1-hydroxy 2-amino 4,6 dinitro benzene) not be made by starting from 2-nitrophenol, reduce the nitrogroup to an amino group and
nitrate the 1-hydroxy 2-amino further to picramic acid?
2-nitrophenol is not probably not very easy to make, as the nitrogroup will most likely be introduced at the 4-postion, so separation is needed one or
the other way, at a certain stage.
Under which circumstances would the nitration take place, probably boiling in H2SO4 and HNO3 for a while?? |
Nitration of 2-aminophenol would put the initial nitro group in the 3- position. Further nitration would probably result in
2-amino-3,5-dinitro-phenol. Note that such products cannot be readily diazotized, since the amino group is in a position where it is
electron-donating to the nitro groups.
For example, 3-nitroanaline can be diazotized with nitrous acid, but not 2- or 4-nitroanaline. And for this same reason, nitration of analine does
not result in 3-nitroanaline.
Nitration with mixed acids on phenol produces about five times as much 2-nitrophenol than 4-nitrophenol.
pure 2-nitrophenol melts at 45degC, whereas 4-nitrophenol melts at 114degC. The two isomers can be separated by steam distillation. Rather, it is
3-nitrophenol that would be very difficult to prepare.
Nitration can be done at room temperature, with mixed acids.
Another bit of information to throw in: benzene is not nitrated by mixed acids at room temperature, but must be heated to boiling,
Interestingly, nitrobenzene can be obtained from the reaction of benzene being boiled with only 65% concentrated nitric acid for about one hour
(reaction is faster if higher acid concentrations are used). The product even shows the presence of a lesser quantity of 2,4-dinitrobenzene, which
becomes somewhat more prevelent after 90 minutes. (I read about this in another forum where a chemistry teacher expressed surprise that the nitration
could be done with such a low concentration of acid)
[Edited on 27-7-2011 by AndersHoveland]
I'm not saying let's go kill all the stupid people...I'm just saying lets remove all the warning labels and let the problem sort itself out.
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by AndersHoveland |
For example, 3-nitroanaline can be diazotized with nitrous acid, but not 2- or 4-nitroanaline. And for this same reason, nitration of analine does
not result in 3-nitroanaline. |
Stil 2-nitrobenzene diazonium perchlorate, 4-nitrobenzene diazonium perchlorate and 2,4-dinitrodiazonium perchlorate do exist...
Also mixed acid nitration of aniline leads mostly to 3-nitroaniline because of the protonation of the amine...to get ortho or para nitroaniline, one
needs to protect the aminogroup from protonation via acetylation for example.
| Quote: Originally posted by AndersHoveland |
Nitration can be done at room temperature, with mixed acids.
Another bit of information to throw in: benzene is not nitrated by mixed acids at room temperature, but must be heated to boiling,
Interestingly, nitrobenzene can be obtained from the reaction of benzene being boiled with only 65% concentrated nitric acid for about one hour
(reaction is faster if higher acid concentrations are used). The product even shows the presence of a lesser quantity of 2,4-dinitrobenzene, which
becomes somewhat more prevelent after 90 minutes. (I read about this in another forum where a chemistry teacher expressed surprise that the nitration
could be done with such a low concentration of acid)
|
I have also noticed that paradox...when all organic chemistry books mention the need to use concentrated HNO3 or mixed HNO3/H2SO4 to allow nitration
of aromatic hydrocarbon.
I have experimentally found that when toluen is subjected to HNO3 (69%) thus with a lot of water, in a closed vessel...it is only a matter of second
before a noticeable yellow colouration occurs at the interface of the toluen layer (on top) and of the nitric water (bottom phase)...this fast turns
into orange and then brown colour (after a few days).
I have taken pictures of that mix now and then initially after hours, then after days, then after monthes...the toluen very soon turns orange, brown,
dark brown...then after a month at ambiant T° (+/- 20°C) thus without any heat or mixing...all the toluen becomes a yellow-orange pale solid...
Following what I have noticed with friends (via solubilisation in NaOH, Na2CO3, and melting points)...it seems to be paranitrobenzoic acid...but I
still have to verify if orthonitrobenzoic acid is absent...it seems very little ortho and paranitrotoluen are also present in the mix...
This is thus a one pot synthesis or paranitrobenzoic acid! Yields remains to
be confirmed. Yields remains to
be confirmed.
[Edited on 28-7-2011 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
Nitration of analine with the usual mixture of concentrated nitric/sulfuric acids is very problematic, because of both charring, and oxidation of the
analine to nitrobenzene
(using a large excess of sulfuric acid helps minimize formation of nitrobenzene). It is true, however, that low yields of 3-nitroanaline (without
significant formation of other isomers) can be obtained, however. Even the use of 60% nitric acid can result in some charring of analine, and this can
be partially reduced by cooling the nitration bath. With 75% concentrated nitric acid added to excess analine, charring takes place fairly quickly.
Adding an acetyl group to the analine, using acetic anhydride, before nitration will make the reaction run much smoother without the byproducts, the
acetyl group later being hydrolyzed off with a mildly strong base, but in this case mostly 4-nitroanaline is produced, with some lesser formation of
2-nitroanaline.
A little known fact is that nitrogen dioxide actually reacts at room temperature (20degC) with toluene to form phenylnitromethane, where a nitro goup
is actually added to the methyl group of toluene. "Phenylnitromethane has been prepared by the nitration of toluene with dilute nitric acid in a
sealed tube."
Konowalow, Ber. 28, 1860 (1895). (the sealed tube probably implies heating) At higher temperatures, addition of two nitro groups on the same carbon
predominates. "nitration of toluene with nitrogen dioxide at a temperature between 20C to 95C yields a mixture of phenylnitromethane and
phenyldinitromethane"
Thus the reaction of the nitric acid with the toluene over a longer period of time might be through the partial decomposition of the nitric acid into
nitrogen dioxide. One would also expect the moderately concentrated nitric acid to very slowly attack/oxidize the toluene, not only producing small
quantities of 2-nitrotoluene, but also other oxidation products together with nitrogen dioxide. The nitrogen dioxide thus formed could then react with
more toluene. Of course, unless the nitric acid was extremely concentrated, the phenylnitromethane would immediately hydrolyze under the acidic
conditions (through a Meyer reaction) to form benzoic acid, the hydroxylamine simultaneously formed would no doubt immediately be oxidized by
additional nitric acid, since at the 60%+ concentrations it is a reactive oxidizer. Fairly complex reaction dynamics. I do not really know what your
orange byproduct is, likely a mix of different compounds, which may possibly include 2-nitrobenzoic acid (this is yellowish-white in color so would
not explain the brownish-orange color).
Nitrous acid (NaNO2 and acid) can be used to nitrate phenol or analine. If concentrated mineral acids are present with the nitrous acid, mostly the
para-nitro will be produced (91% yield), but if glacial acetic acid (concentrated) is present with the nitrous acid, yields of 74% ortho-nitro are
possible. Ortho means the nitro is in the 2-position, adjacent to whatever other group is on the ring. Para means the nitro is on the opposite end of
the ring from the other group.
The theory is that a nitrosyl ion (NO+) initially nitrates the ring, and then the resulting nitroso group gets oxidized to a nitro. ("Aromatic
nitration", K. Schofield. 1980)
However, this nitrosyl ion equilibrium with nitrous acid is only present under strongly acidic conditions, certainly not when nitrous acid is in an
alkaline envirorment., where it is mostly in the form of nitrite.
One source mentions that reaction of analine with nitrosonium hydrogen sulfate NO[+] [-]SO4H produces nitrobenzenediazonium salts.
NO2C6H4–N[+]ΞN
First, the nitro group is added to the analine. It is only after the acids have been somewhat diluted with water that the amino group can be
diazotized, as diazotization cannot take place in extremely concentrated sulfuric acid.
"Interaction of Analine with diazotizing and nitrating Agents in Concentrated sulfuric Acid"
Mikhail V. Gorelik, Vera I. Lomzakova, Elena A. Khamidova
Research Institute of Organic Intermediates and Dyes, Moscow, Russian Federation
nitrosonium hydrogen sulfate is the same thing as nitrosyl sulfuric acid, which may be prepared by bubbling mixed nitric oxide/nitrogen dioxide gases
into concentrated sulfuric acid.
(3)H2SO4 + NO2 + NO --> (2)NO[+]SO4H[-] + H3O[+]SO4H[-]
Whereas 3-nitroanaline can be easily diazotized with dilute nitrous acid, both 2-nitroanaline and 3,5-dinitroanaline require moderately concentrated
sulfuric acid to be present, for formation of nitrosonium ions.
Treatment of a 1:1 ratio of nitrosylsulfuric acid to analine, using 92.5% concentrated sulfuric acid, leads to formation of about three times as much
4-nitrodiazonium salt as 3-nitrodiazonium salt. Using 95.5% sulfuric acid leads to a 6 to 4 ratio of the isomers, while 100% concentration leads to
equal formation of both. The treatment of previously prepared benzenediazonium salt with nitric acid does add any nitro group.
[Edited on 28-7-2011 by AndersHoveland]
I'm not saying let's go kill all the stupid people...I'm just saying lets remove all the warning labels and let the problem sort itself out.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
The first three pictures are of the picramic acid produced which was described in the "picramic acid from picric" thread. The last three pictures are
of the DDNP produced from it. I used the method from COPAE to perform the diazotization reaction and produce DDNP. It is a very simple procedure. Once
the picramic acid is obtained it is really all downhill from there.
The product is drying right at the moment, but I will be doing some testing soon. It can be recrystallized from acetone, by pouring ice cold water
into an acetone solution of it according to several sources.
Several people have discussed DDNP performance issues with respect to density. I personally believe at this point that lousey reducer type and purity
has lead to low quality and low purity DDNP in most cases. However, I did find this in TM 9-1300-214 ("Military Explosives"):
"If this material is dissolved in hot acetone and a large volume of ice water added to the agitated solution, the DDNP is precipitated as a bright
yellow, amorphous powder. Recrystallization from a solvent is used to produce the tabular crystals that comprise specific grade material."
 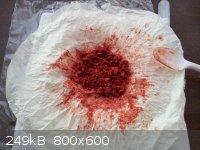    
The reason the product is so clumpy is because it was squeezed very hard in paper towel in order to blot away as much water as possible after
filtering.
[Edited on 28-1-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Well, the DDNP is nice and dry this morning. Yield was quantitative based on picramic acid. I think this is the first time I have ever gotten a
quantitative yield for any synthesis. I lit a little and it flashed and made a little whomp sound kind of like mercury fulminate (but without the
black/grey residue). I put about a paper match heads amount in a little aluminum foil, placed it on a concrete floor, and gave it a little wrap with a
hammer which detonated it very easily. Now my ears are ringing of course. I said
I was going to stop doing stuff like that, since I have noticed my hearing has been getting worse these last few years.
I am going to be in a more rural area in the next couple of days, so I will be testing its ability as an initiator of secondary explosives.
[Edited on 28-1-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
DDNP Recrystallization
I have been thinking about how best to grow larger more well defined crystals of DDNP in order to improve the density and performance. Most of the
common solvents for DDNP don't really have large enough differences in their ability to dissolve DDNP going from cold to hot solvent temperatures.
After thinking about it a little bit I believe that slow, controlled, evaporation is a method which would probably be very effective for growing the
large well formed crystals of DDNP that are desired. This process is not, however, a purification process. For purification purposes, the method
involving pouring ice cold water into a nearly saturated solution in acetone could be used, if purification was desired first.
The following website does a very good job of explaining recrystallization, and crystal growing concepts, including slow evaporation.
http://www.chemistryviews.org/details/education/2538901/Tips...
[Edited on 28-1-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
| Pages:
1
2
3
..
33 |













