| Pages:
1
..
26
27
28
29
30
..
33 |
nitro-genes
International Hazard

Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Hypochlorite mediated oxidation of 2,3,6 trinitro-4-aminophenol to a furoxan might be tricky due to the ease of hydrolysis of any possible
quinone-imine formed under these conditions due to OH/NH2 group in para, likely formation of chloropicrin from nitrophenols in general and the well
described incompatibility of the compound with bases, decomposing to form tarry products.
Going through the excerpts posted, it seems any possible furazan/furoxan from the nitration of isopicramic acid could be harder to distinguish from a
diazophenol than I thought, potentially sharing similar melting points, colour, explosion temp, solubility in strong acids and light sensitivity. 
I wondered if the trinitrodiazophenol obtained from the nitration of isopicramic acid may rearrange in water or under slightly basic conditions to a
benzofurazan/furoxan as opposed to only displacement of the 3-nitro by water or OH-, but can't figure out a likely mechanism, since it would entail a
series of complex rearrangements not generally known for diazo(nium) groups and unlikely maybe due to the para orientation of the OH/NH2 group.
Couldn't find any examples of furazan/furoxan formation from diazonium precursors in literature at least, except via azide substitution of the diazo
group and oxido-redox with an ortho nitrogroup, like in the synthesis of KDNP, which probably doesn't occur through a quinone-imine precursor in the
first place. The latter also suggests that any furoxan formed wouldn't react further with the excess azide used in some reaction schemes outlined,
which directly contradicts the observed formation of nitrogen for the compound obtained from further nitration of isopicramic and seems the best
argument the compound truly is a diazophenol. Moreover, the in literature described "boiling dilute SA-excess nitrite treatment" of mono and
dinitro-4-amino resorcinol derivatives also results in a compound that forms salts, which would be hard to explain as a furazan/furoxan without an
extra nitro group present ortho relative to the amino group after nitration, which seems very unlikely to occur under these conditions.
As an interesting side note, I subjected 2-nitro 4-diazophenol to the "boiling 30% SA-excess nitrite" treatment and obtained a product which proved
very similar to pDDNP with regard to oxygen balance, so definitely a second nitro group was introduced. Where was it likely to end up? Position 3,5 or
6? 
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The acetaminophen derivative I thought likely to form a possible phenolic analogue of KDNBF would be the 3-nitro derivative of paracetamol acetate
where the ortho related 3-nitro and 4-amino (after deacetylation) would provide the structure susceptible to oxidation by hypochlorite as occurs for
the o-nitroaniline precursor oxidized to benzofuroxan and further nitrated for KDNBF.
If there is possible a HO-DNBF it would be somewhat analogous to the HO-DDNP or DDNR, only it should be a diacid. When a hydroxyl is added to the
ring for DDNP or p-DDNP it results in a better oxygen balance and increased energy, so it seems that could be true also when a hydroxyl is added to
the ring of DNBF.
Use of acetic anhydride to form paracetamol acetate directs the first nitro group to 3 and forms the needed ortho relation of nitro and amino as would
occur for the o-nitroaniline precursor for KDNBF.
[Edited on 1/9/2018 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
@nitro-genes
Reviewing some of the earlier research hasn't been done so I am going on memory but there was a Japanese Pharmaceutical Bulletin article that
misidentified a 3-nitro derivative of paracetamol, that was instead actually a 3-nitroso. If they got the structure correct for the ring position 3,
then it seems possible a nitroso at 3 could be attached first and then the 3-nitroso could be converted to a 3-nitro. It may be that this was
discussed at the earlier time and ruled out for some reason. I recall there were issues with duplication of that reported synthesis, but have wondered
if a conventional nitrosation approach would not also introduce a nitroso at 3 on paracetamol. If it does work then that could be used to avoid the
requirement for acetic anhydride to form paracetamol acetate and get selective reaction on position 3 for the entering group.
Position 3 nitration is the key pathway for DDNR and iso-DDNR and for the speculative hydroxy KDNBF if it exists.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
So here a follow up of the conversion of picric acid to picramic acid using copper(II)sulfate and ascorbic acid from the short questions quick answer
thread discussion with Rosco Bodine:
After some experimentation, it seems no necessity to increase the pH during the reaction at all.  The entire reaction seems to be facilitated by ligand coordination of ascorbic acid by the copper(I) or (II) towards
the picric acid, resulting in a very high specificity of the reaction, resulting in the reduction of 1 ortho nitro group of picric acid, producing
picramic acid, which eventually seems to precipitate during the reaction as the insoluble copper(I) or copper(II) salt. The first step probably
comprises forming a complex of Cu(I/II)(Ascorbic)x(Picrate), which upon heating for some time produces copper picramate. The entire reaction seems to be facilitated by ligand coordination of ascorbic acid by the copper(I) or (II) towards
the picric acid, resulting in a very high specificity of the reaction, resulting in the reduction of 1 ortho nitro group of picric acid, producing
picramic acid, which eventually seems to precipitate during the reaction as the insoluble copper(I) or copper(II) salt. The first step probably
comprises forming a complex of Cu(I/II)(Ascorbic)x(Picrate), which upon heating for some time produces copper picramate.
https://books.google.nl/books?id=PshJAQAAMAAJ&pg=PA407&a...
Experimental 1:
1 gram of picric was added to 25 ml distilled water and brought to near boil. The picric solution was adjusted to a pH of 7 using 10% NaOH solution,
then 1.1 grams of copper(II)sulfate pentahydrate (~1 molar eqvt relative to picric) was added at once. When all had dissolved into a greenish
solution, 2.7 gram ascorbic acid (~3.5 molar eqvts) were added at once. The solution went a clear dirty greenish-yellow-brown first. After a few
minutes of heating, large amounts of a greenish precipitate started to develop. The solution was kept near boil for another 15 minutes, which seemed
to change the colour to a yellow-greenish (not sure yet the extended boiling time is necessary at all). During heating, lots of an odorless gas was
produced, evident by a lot of foaming. Most of this had died down after the 15 minutes of heating. The solution was cooled and the yellow-greenish
precipitate filtered off, which after drying weighed 0.77 grams (~67% yield, assuming pure copper picramate). 25 mls of 10% HCl was gently evaporated
down to 10 ml. The greenish precipitate was added in small amounts untill no more was able to dissolve. Slowly the solution was diluted with distilled
water which precipitated small needles of an orange-red compound. After washing thoroughly, this was diazotized at 0 deg C., producing a bright yellow
compound that flashes a lot like DDNP.
An experiment looking at the catalytic potential of copper salts was performed earlier.
Experimental 2:
1. 0.25 grams of picric were added to 10 ml water and brought to 80 deg C. Then, 0.6 grams of ascorbic acid were added, which produced no obvious
colour change. Using NaOH solution, the picric/ascorbic was adjusted to a pH of around 7, which also did not result in any significant change in
colour within 5 minutes. A few crystals of copper(II)sulfate (~5 mg) were added, which immediately produced a clear solution changing rapidly in
colour, going from yellow, to orange, to dark orange red, finally resulting in a fine orange-red precipitate at the bottom of the beaker (Seemingly
too much to be explained by the copper addition alone, though would need to be weighed). Out of curiosity, another 0.6 grams ascorbic acid was added
in small increments (everytime adjusting the solution again to pH7. Strangely, after only a few spatules of additional ascorbic, the solution began
producing large amounts of some odourless gas, foaming significantly and changing to a very dark red-orange colour, with only a small amount of
precipitate, that most likely is copper powder. The dark red-orange solution was acidified using sulfuric acid, resulting in a much lighter orange
solution and a precipitate of brownish crystals, that on closer examination proved to be unreacted picric acid.
2. 0.6 grams of ascorbic acid was dissolved in 10 ml water and brought to around 80 deg C.. Then added a few crystals of copper(II) sulfate, there was
no colour change. Then adjusted the solution to pH of 7, which produced a slightly opaque solution with slight orange sheen (Probably copper(I)
oxide). A spatule of salicylic acid was added, resulting in no colour change. Finally, a spatule of picric was added, immediately going from orange to
dark orange-red again.
It seems from the latter experiment that copper salts are truly increasing the reduction rate at a pH of 7. Very interesting, the question is what
formed here, how specific the reduction is and how the conversion efficiency is (if it can truly acts as a catalyst) Probably not since presumably
conversion to copper powder is also happening at this pH and temperature, explaining the unreacted picric recovered.
This may well be the weirdest way to produce picramic acid, though it is not entirely sure yet the compounds isolated are truly picramic and DDNP, a
dinitro nitroso phenol could behave similar energetic maybe. Also not entirely
sure what the bright orange-red compound is, presumably picramic, though the melting point seems to be on the low side. I've wondered if a picramic
picrate salt could exist, since picric itself is a very strong acid and what it's properties would be. Thorougly washing the DDNP produced with warm
water and measuring yield could provide an answer to that. Alternatively, maybe dehydroascorbic could condense with the picramic produced or
something, which on diazotization reverts back to DDNP.
All in all, the ligand orientation mechanism seems most plausible for the reduction, since catalytic amounts of copper do not seem to have the same
results till now, although this would need more experimentation. Alternatively the reduction may be catalyzed by copper(I) itself somehow.
[Edited on 1-2-2018 by nitro-genes]
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
Has the reduction to picramic acid ever been considered with iron via the Bechamp reduction? The way I see it is dissolving the picric acid in an
organic solvent. Adding the iron powder and introducing the HCl slowly. Then reflux until the iron dissolves. Finally NaOH is added to precipitate
iron hydroxide. But that will also produce sodium picramate so the solvent would need to keep that in solution until the iron hydroxide sludge was
removed. Then a vacuum distillation to separate the sodium picramate from the solvent. Any ideas if it's feasible?
https://en.chem-station.com/reactions-2/2017/05/bechamp-redu...
This link has a similar reaction with an aryl amine.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
IIRC, Axt posted a reference long time ago regarding the reduction of picric to picramic using just iron and water at 80 deg C. (somewhere in the
picramic thread) and I think Rosco posted a brief reference regarding reduction using zinc metal/ammonia in boiling methanol for half an hour, so it
might be possible.
No chemist myself and the specificity and precise reactions occuring during these different nitro reductions are still largely a mystery to me, so
sorry if this seems like a load of unscientific crap:
Bechamp seems to work fine for most mono nitro arenes, though I thought in aquous solutions at least, there is no specificity for the reduction of a
specific nitrogroup in the case of polynitroarenes. Not sure why this is though, maybe due to proton-tranfer or hydrolysis reactions, there seem to be
2 independ pathways during nitro reduction, one via direct reduction and one via condensations of partially reduced intermediates, maybe this is one
of the reasons?
http://pubs.rsc.org/-/content/articlelanding/2014/cp/c4cp043...
In anhydrous solvents it seems possible to selectively reduce 1 single nitrogroup using nascent hydrogen, for example 1 nitrogroup of TNT can be
reduced specifically using gAA/Fe(0). Since I lack any glacial acetic, I tried the reduction of TNT in ethanol/HCl/steelwool once, which seemed to
work in producing 2-amino 4,6-dinitrotoluene (although not very pure). If the same could work for polynitrophenols...no idea...might be possible...
one thing that might be different for nitrophenols is that the OH group itself may also form salts (that may precipitate from most OTC solvents for
example) and maybe also change the way partially reduced intermediates would behave during the reduction, for example a nitrosophenol would be in
equilibrium with a quinone-oxime. Maybe the reactions occuring may also largely depend on the pH and solvent used, so I wonder if one set of
conditions would apply to different nitro arenes anyway.
Also did another experiment with the Cu(II)/ascorbic acid reduction of picric again. After adding the ascorbic acid to the copper sulfate/sodium
picrate solution, the greenish precipitate was immediately filtered off and thoroughly washed with cold water after it formed (so without any further
heating applied). It dissolves in 10% HCl easily, but only picric can be isolated from this and not a trace of picramic acid. So it seems likely a
complex of ascorbic/Cu(I) or Cu(II) and picric is formed first which goes through some internal reduction forming picramic acid. Very interesting,
although maybe not economical, never seen picramic of this purity before. It would indeed also be interesting to see if iron(II) instead of Cu(II)
would behave similar under these conditions.
I'd like to scale this up a bit to be able to measure yields etc, though I'm hesitant due to the odourless gas produced during heating. My first guess
would be carbon dioxide from decarboxylation of some dehydroascorbic acid derivative, but not sure if it could be something toxic as well (carbon
monoxide, or some organocopper compound).
"Crystal structure of a copper complex of 2-carboxypentonic acid; A decomposition product of dehydroascorbic acid (DOI10.1039/dt9870002905)
Abstract
In acidic aqueous solution and in the presence of copper(II), ascorbic acid is rapidly oxidized to dehydroascorbic acid, which rearranges to give the
branched-chain dicarboxylic acid 2-carboxypentonic acid (1,2,3,4-tetrahydroxybutane-1,1-dicarboxylic acid)(H3cpa). The ion cpa3– is sequestered by
copper(II) to produce the insoluble crystalline product [Cu9Cl2(cpa)6(H2O)3]2–·xH2O"
Any ideas what kind of gasses could be produced? Is gas production/foaming also observed during the making of nano copper from Cu(II) and ascorbic for
example?
[edit] What do you know, an OTC source of copper(II)sulfate! People actually just pour this down the drain?
https://www.doitbest.com/products/477796
[Edited on 4-2-2018 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
After some experimentation it seems possible to use copper salts as a catalyst for the reduction of picric acid to picramic acid. Between 0.0001 and
1.1 molar equivalents (relative to picric) of a salt containing the Cu(II) or Cu(I) ion can be used as a catalyst for the reduction of picric acid (or
any of its picrate salts) solutions in water to form picramic acid or any of its picramate salts in high purity and yield. The reaction can be
performed at a temperature of 20 deg C. to 100 deg. Celcius, (preferentially between 60 and 90 deg Celcius), and at a pH of about 2-12 and using a
variety of reducing agents, including ascorbic acid, glucose (and other reducing sugars), sulfites and other common reducing agents known in the art,
for which between 1.5 to 6 molars are preferentially used for each mole of picric. Depending on the reducing agent and temperature utilized, reaction
times can vary from 5 minutes to 8 hours. Depending on the reducing agent utilized, an aquous solution of the reducing agent is preferentially added
to an aquous solution of picric acid or any of its picrate salts, in other reactions the reducing agent is directly added to the picric acid or
picrate solution, and the catalyst solution containing Cu(II) or Cu(I) salts is gradually added over timeperiod of the entire reaction. The picramic
acid obtained is of exceptional purity and can be directly used to produce diazodinitrophenol (DDNP) by diazotization using 5-20% HCl and a nitrite
salt at 0-5 deg. C.. The light yellow DDNP thus produced is obtained as spherical crystal agglomerates of high bulk density, that can be directly
used for primer applications.
It would also be interesting to see whether something like styphnic acid can similarly be reduced. 
[Edited on 5-2-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
If you are getting picramic acid from an acidic reaction mixture down to a pH of 2, that is very interesting and contradicts a lot of literature that
reports an alkaline condition picrate as a requirement. Maybe it is only the presence of some alkaline picrate in part as a component in a partially
neutralized picric acid reaction system that is required, and the pH for the reduction mixture of a "partially neutralized" reaction system may still
be acidic and not interfere with reduction of that component that is already neutralized. Glycine can be used to greatly increase the solubility of
picric acid in an excess of picric acid required for formation of Diglycine Picrate, which would be an acidic mixture or greatly more soluble and
concentrated picric acid. Glycine also complexes copper so this could be an interesting material for experiments as a possible additive that could
possibly make the catalytic copper salt even more active. The copper glycine complex is reportedly not stable above 90 C so that factor should rule
out use of an excessively hot reduction mixture. See attached articles.
Information from the articles indicates that the glycine cobalt complex could also be a candidate regenerable catalyst for reduction. Reportedly the
Glycine Cobalt Picrate itself is not unstable to boiling heat. It is unknown how soluble is the different lower oxidation state cobalt glycine
picrate or how soluble or reactive are either oxidation state in reaction with ascorbic acid or ascorbate. Here what is contemplated is that the
soluble glycine cobalt picrate itself could possible function as a regenerable catalyst which could act as a reducing agent on the remaining 99% bulk
of the picric acid or soluble picrate salt such as the sodium or magnesium salt.
The discussion posts related to reduction schemes for picric acid / picrates to picramic acid / picramate should be exported to the dedicated thread
for the topic to keep the subject matter in one place. Here is a post link for that thread. Historical references being researched on some of these
niche topics have for years been resulting in Google search hits for this science forum as a kind of repository for "obscure and interesting"
collected references and experiments so we should keep things organized.
http://www.sciencemadness.org/talk/viewthread.php?tid=433&am...
Ferrous ammonium sulfate Mohr's Salt is another likely candidate for a reducing reagent, the double salt or complex of Ferrous Sulfate and Ammonium
Sulfate, which upon oxidation to Ferric Ammonium Sulfate expels one Ammonia. References to "the ammoniacal liquid" without further details in
historical references to reductions involving iron indicate that ammonia was used as a base but do not specify a step by step or reaction specifics to
know exactly how ammonia was used.
A second reducing agent like ascorbic acid / ascorbate, or a sulfide added gradually can be used to regenerate the "ous" lower oxidation state iron or
copper or other transition metal salt, from its oxidized "ic" higher oxidized state so it acts as a regenerable catalyst and the reduction continues
so long as the catalytic salt does not further react to form some inactive byproduct, or precipitate as an insoluble oxide, which in effect would
"poison" the catalyst and require more of it to be added to continue the reduction.
Glycine is something that has caught my attention as possibly useful as a chelating agent that has potential value because glycine is also reactive
with picric acid, forming diglycine picrate and increasing the solubility of picric acid by that association, particularly in solution where the
picric acid is in excess.
Glycine also complexes copper and is known to inhibit the precipitation of some metal hydroxides up to an alkaline condition of 10.3 pH. Because of
the activity of glycine it could have usefulness as a protector of the catalyst by inhibiting precipitation of an inactive byproduct and counter the
tendency of the catalyst to be deactivated by undesired further reaction. The glycine could also serve as a buffer for the pH or as part of a
buffering scheme in mixture with other salts of possible benefit also.
There is probably an optimum process scheme that varies somewhat for the particular transition metal salt being used. Solubility of the reactants is
an important factor and greater solubility is generally better because more concentrated solutions generally react more quickly and completely.
Attachment: diglycine picrate J. Biol. Chem.-1912-Levene-285-94.pdf (554kB)
This file has been downloaded 560 times
Attachment: glycine picrate related J. Biol. Chem.-1958-Selim-157-62.pdf (271kB)
This file has been downloaded 611 times
[Edited on 2/5/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Supposedly, the pKa of picric, ascorbic and picramic are 0.42, 4.17 and about 4 respectively. So despite a pH of about 3 after the ascorbic addition,
IIRC, more than 99% of the picric should still be present as sodium picrate indeed. My guess would be that the ascorbic is acting as a ligand best at
its "native" pH, which is not acidic enough probably to interfere with the precipitation of the insoluble copper picramate (which is soluble in strong
acids).
There remains to be a lot determined regarding this reaction it seems, other ligands like glycine could help with the reduction or not. They may keep
the copper(I) and copper(II) soluble during the reaction, but may also precipitate as a copper(glycine)picrate or picramate or not increase the
solubility of the copper picramate or just interefere with ligand coordination of the ascorbic by competion with ascorbic in general, as also seems
the case when using ammonia.
There is a lot to this reaction it seems and would need to be determined:
1. What is the green complex formed initially in the reaction?
2. What is the yellow-greenish precipitate after the reaction is completed? A copper(I)picramate, copper(II)picramate, a complex of a copper picramate
and some dehydroascorbic product, a condensation product?
3. Decomposition temperature of copper(picramate) (prolanged boiling will degrade it supposedly)
[Edited on 7-2-2018 by nitro-genes]
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
Quote: Originally posted by nitro-genes  | After some experimentation it seems possible to use copper salts as a catalyst for the reduction of picric acid to picramic acid. Between 0.0001 and
1.1 molar equivalents (relative to picric) of a salt containing the Cu(II) or Cu(I) ion can be used as a catalyst for the reduction of picric acid (or
any of its picrate salts) solutions in water to form picramic acid or any of its picramate salts in high purity and yield. The reaction can be
performed at a temperature of 20 deg C. to 100 deg. Celcius, (preferentially between 60 and 90 deg Celcius), and at a pH of about 2-12 and using a
variety of reducing agents, including ascorbic acid, glucose (and other reducing sugars), sulfites and other common reducing agents known in the art,
for which between 1.5 to 6 molars are preferentially used for each mole of picric. Depending on the reducing agent and temperature utilized, reaction
times can vary from 5 minutes to 8 hours. Depending on the reducing agent utilized, an aquous solution of the reducing agent is preferentially added
to an aquous solution of picric acid or any of its picrate salts, in other reactions the reducing agent is directly added to the picric acid or
picrate solution, and the catalyst solution containing Cu(II) or Cu(I) salts is gradually added over timeperiod of the entire reaction. The picramic
acid obtained is of exceptional purity and can be directly used to produce diazodinitrophenol (DDNP) by diazotization using 5-20% HCl and a nitrite
salt at 0-5 deg. C.. The light yellow DDNP thus produced is obtained as spherical crystal agglomerates of high bulk density, that can be directly
used for primer applications.
[Edited on 5-2-2018 by nitro-genes] |
Are you quoting a patent or something here?
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Did some experiments this weekend:
First I tried to determine the minimal temperature for the reduction on a very small scale using 1 mole eqvt of Cu(II)sulfate and 3.1 mole eqvts of
ascorbic acid. At 40 deg C., the complete formation of the initial green precipitate (as evident by a nearly colourless filtrate) takes about 15
minutes. At 40 C, there is hardly any reduction going on and the precipitate remains bright green if kept at this temp for 60 minutes. The temperature
was then raised gradually to around 60 deg C., gas formation became evident and within another 20 minutes or so the precipitate attained a more
yellowish colour. So 60 C. seems minimal for the redution to take place and is concomitant with the gas formation.
The gas is probably carbon dioxide, resulting from oxidation of ascorbic to dehydroascorbic, which hydrolyses to 2,3 diketo gulanic acid, which
decarboxylates further to various other products (attachment), which probably explains the brown colour obtained from prolonged heating of the
reaction.
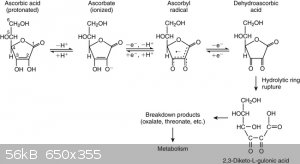
Also did the reduction on a slightly larger scale, determining yield of the picramic obtained and tried the reaction with 1 and 0.5 molar eqvts of
copper(II)sulfate to try and minimize the amount of copper salts needed.
Exactly the same reaction conditions were used:
2.3 g picric was suspended in 25 ml dH2O and warmed to 60 deg C. A 10% NaOH solution was gradually added to a pH of 7. When done slowly, no pH paper
is needed, the picric itself functions as a reliable indicator of pH. Upon each addition of the NaOH, an orange colour is observed, which quickly
reverts back to a bright yellow again when neutralized by the undissolved picric. When all the picric has dissolved, a few extra drops of NaOH are
added until a permanent slight orange colour is obtained. Then Cu(II)sulfate pentahydrate was added (2.5 g in Exp 1 vs 1.25 g in Exp 2), which
dissolved into a clear bright green solution. Then 5.46 g (3.1 molar eqvt) of ascorbic acid was added. After a few minutes or so, an abundant bright
green precipitate started to form and a mild exotherm could be observed. The solution climbed to 70-75 deg C on its own and was kept there for 30
minutes. The precipitate changed to a yellow-greenish colour, was filtered and washed 3 times using cold water.
After weighing and drying, the precipitate was dissolved in the least amount of boiling 20.2% HCl, (about 15 ml). Upon dilution with icecold water to
100 ml, small bright orange-red needles of putative picramic acid were obtained, which were washed, dried and weighed. The filtrates were saved and
copper(II)oxide can be retrieved from them by basifying with NaOH and boiling.
Results:
Exp 1: 2.22 g of putative copper (I/II) picramate, 1.32 g picramic acid (66.3% yield).
Exp 2: 1.65 g of putative copper (I/II) picramate, 1.00 g picramic acid (50.2% yield).
The filtrate of the reduction experiment 1 was saved and a dark brown in colour (probably from dehydroascorbic decomposition products), upon
basifiying with NaOH, the solution became a very dark red colour (maybe some diamino nitrophenol) and some residual Cu(I)oxide was present as a light
yellow precipitate.
Any ideas how to maximize the yield further? In both cases, after 30 minutes into the reduction, there was still lots of gass formation. Maybe it just
needs longer reaction time? Not sure if the decarboxylation observed is a necessity for the reaction to proceed and is a direct indicator of the
formation of dehydroascorbic acid, which probably leaves the complex and allows another ascorbic acid to complex. Depending on the stability of the
copper picramate, maybe keeping at 60 C. until no more decarboxylation is observed results in higher yield. Maybe larger dilution of the reaction also
helps.
[Edited on 11-2-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Considering that the picramate of copper is insoluble it would rule out that copper can be used as a regenerable catalyst because ALL of the copper in
equimolar proportion is subject to sequestration as insoluble picramate byproduct. The copper has catalytic activity with respect to the ascorbic acid
and to keep shifting the equilibrium for the reduction by precipitation of the picramate as a cuprous salt. But in terms of efficiency as a catalyst
to be regenerated, iron or manganese or cobalt or nickel salts would be better choices in terms of potential efficiency for the reduction, because the
picramates for those metals should not precipitate as occurs for the copper picramate.
Girard, Comptes Rendus, March 7, 1853 pg. 421
If your product of reduction is cuprous picramate then half of the reducing power of the copper is being sequestered in the product.
At the end of the reduction reaction, copper values in the spent reaction system would desirably be the +II cupric salts, but that may not be the case
if the precipitate is instead the cuprous picramate. It may be unavoidable that it happens that way if that is what is occurring and changing the pH
to more alkaline may or may not help. This reaction involves a set of factors that are variables difficult to predict what reaction wins out, so that
conditions can be optimized. Guesses and experiments are ahead.
Suggest to increase the amount of CuSO4 150% of the 2.5 g of Experiment 1 and compare and then a second experiment with the same 150% increase applied
to the ascorbic acid as well. As a variation the added amount of CuSO4 could be mixed with the ascorbic acid and the mixed reagent added gradually as
a 3rd experiment. Adjusting the pH conditions of the reducing reagent solution, or adjusting the pH of the picrate reaction mixture with a buffer like
magnesium oxide in suspension in the stirred mixture is another idea. The addition of a chelating agent like glycine is another idea for possibly
increasing the efficiency of the copper catalyst which appears to be tied up as a reagent as much as it is acting as a catalyst. It seems it is
capable of acting as a catalyst but is also being consumed as a reagent, and sequestered as a byproduct, so that an additional available quantity that
can regenerate may improve the yield.
The reducing "power" of ascorbic acid is greatest for the free acid at about pH 2 and between 2 and 4.1 is the most active range if what I seen
reported is accurate. In that acidic range it appears the reducing power is effectively double what is the reducing power for ascorbate ion in an
alkaline pH range, where half the available reducing hydrogen is already consumed in association with a base like sodium if the ascorbate being used
is sodium ascorbate. The available reducing hydrogen reduction "capacity" then exists for a first order "mono"-dehydroascorbate intermediate that is
further reduced to dehydroascorbate which has formed from a loss of TWO reducing hydrogens with respect to ascorbic acid. Evidently the distinction
between "protonation" by an acid hydrogen and an added hydrogen by "reduction" is a variable that depends entirely on pH. Evidently in an acidic pH
reduction reaction system a +2H reducing power exists for free ascorbic acid. At intermediate pH there is a +1H reducing "potential" and at a strongly
alkaline pH there is a zero reducing potential with the ascorbic acid appearing as an inactive di-acid salt.
The pH factor is not conclusively identified to be strongly alkaline for the reduction reaction system of where picric acid / picrate is the target
for reduction, and there may be a tradeoff for the best reaction system where a slightly acidic reduction mixture benefits increased reducing power
for ascorbic acid, but an increased alkalinity may benefit the reduction and selectivity for production of the target picramate. Somewhere is an
intermediate range of pH and temperature and concentration that "just works" best for the
process using a combination of reagents. Working out what is that optimum combination for the best result is a lot of trial and error until it gets
"dialed in" what is the "secret formula" that works best.
Have you been curious enough to do a glowing splint test for oxygen on the gas?
I have a suspicion your colorless gas evolution may be oxygen. And if it is oxygen then it
will take a lot less ascorbic acid for the reduction according to theory than would be required for reduction by a sulfide. My suspicion is that the
ascorbic acid is manifesting the surgical precision of an enzyme, with one or a pair of ascorbate ions operating directly on the N=O2 group and
cleaving, splitting that radical without actually combining with the freed oxygen, attaching an H or H2 hydrogens directly to the N to form the (di-)
hydrogenated N, (NH2) amino radical. It would be a small wonder if that is indeed the mechanism occurring.
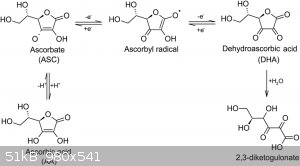


Attachment: Chemical_Gazette 1853 article Aime Girard picramic acid.pdf (284kB)
This file has been downloaded 571 times
After reviewing the descriptions for pH affecting ascorbic acid, and noting the insolubility of copper picramate, it seems that use of copper may be
better applied in the isolation and purification step where a soluble picramate in solution with other reduction reaction byproducts is mixed with a
soluble copper salt to precipitate the insoluble copper picramate filtered out to isolate the picramic acid value as the insoluble copper salt, washed
and redissolved in an acid to resolve the solution into its separated components with free picramic acid precipitated from a solution of the acid salt
of copper that remains dissolved.
Magnesium seems to be the most likely candidate for a picrate to be reduced by ascorbic acid due to the high solubility of the magnesium salt and the
limited alkalinity which would be inherent due to the low solubility of magnesium hydroxide and the limit of a 10.3 pH that would occur for a solution
of magnesium hydroxide alone. A solution of magnesium picrate would be much closer to neutral, and would be an excellent target in the pH range
"sweet spot" for reduction by free ascorbic acid alone or partially or fully converted to a sodium or magnesium ascorbate. HCl would be cheap acid
that should work fine for adjusting pH for the reduction or for the later isolation and purification, that could probably done with or without using a
copper salt to obtain an insoluble picramate intermediate.
Also still of interest is something I mentioned before about the reported increased solubility of picric acid associated with glycine to form
diglycine picrate with the picric acid in excess to possibly more equivalent what would be a mono-glycine picrate. The pH of such a mixture would
definitely be acidic and might be susceptible to reduction by ascorbic acid directly, and may precipitate free picramic acid directly as fast as it
forms ....if there is not a similar association with glycine causing increased solubility for picramic acid....then the free picramic acid should
precipitate directly in pure form, from an efficient ascorbic acid reduction that may be optimized in the acidic pH reduction scheme. This is
theoretical and would depend on the selectivity for reduction to picramic acid and would it proceed at an acidic pH similarly as occurs in the
experiment where a copper salt is used. It would definitely be an experiment worth trying since it could be exactly the reaction condition that is
desirable if it does work as anticipated. It could be a model lab method for easy synthesis of pure picramic acid as a one pot synthesis, and may
possibly even be extended further to a one pot synthesis of DDNP.
What I am thinking about as an extension even further of the "one pot" reaction scheme possible is the salicylic acid or possibly aspirin starting
material which is first sulfonated and then under mild conditions in dilute solution nitrosated and nitrated to picric acid quantitatively....not even
isolating the picric acid...add glycine and proceed with an ascorbic acid addition, followed by sodium nitrite or the variant for diazotization using
a nitrate and copper wire. DDNP could possibly be obtained by a much simpler path of reactions under milder conditions than the more usual and better
known methods.
[Edited on 2/12/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Justed tested on a very small scale a near boiling solution of picric acid itself (without any neutralization) with 3.1 molar equivalents of ascorbic
and 1 molar equivalent of copper(II)sulfate. To my surprise, a yellow-greenish precipitate formed, from which (by dissolving in 20.2% HCl, the orange
compound (presumably picramic) precipitated on dilution with water in what appears to be a decent yield at least. I was curious if at this low a pH,
the putative copper picramate would be able to keep partially dissolved during the reaction and allow the copper to act as a catalyst. Tested this
using 0.05 molar eqvts of Cu(II)sulfate, but although the solution goes to a dark orange-red-brown very quickly at 70-80 deg C after adding the
coppersulfate, only some black crystaline solid and some copper metal seemed to have formed after 2 hours, both of which won't dissolve in HCl.
[Edited on 13-2-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Ascorbic acid seems to be the exception to the widely "accepted" narrative in the literature about reduction conditions for producing picramic acid,
all of which references I have ever seen are reporting alkaline condition as a requirement. Once again it seems the highlighter is being applied to
"textbook" generalizations that have a SM footnote inserted regarding a HUGE exception to the "rule" having been identified by this science forum. Go
figure. So much for academic preeminence "authority" meeting up with test tube anarchists leading a molecule rebellion. It's a veritable ionic uprising.
Joking aside....Ascorbic acid is reportedly heat sensitive which is why cooking foods have reduced vitamin C levels compared to raw uncooked food. So
even though the activity and reaction rate increases with temperature, there is a half life for ascorbic acid that shortens with elevated temperature,
and it is not heat stable. I'm not sure what that parameter is exactly. Also it occurs that most of the studies about ascorbic acid are in vivo and
there can be different behavior in vitro. So there is not necessarily a specific rule that what happens in a biological reaction system also happens
the exact same way in a laboratory synthesis. In a living organism other enzymes interact with ascorbic acid to do various things so isolating what
occurs for ascorbic acid in a reaction in a flask is a different kettle of fish.
[Edited on 2/13/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Are basic conditions really a necessity for the reduction of picric to picramic? What would happen if a solution of picric acid in gAA or other
solvent would be reduced with H2S, stannous chloride, zinc or iron powder e.g., never seen any references for this at least. My uneducated thinking
(without much reading) was that the impurities arising from the sulfide reduction are actually due to the high pH employed during the reaction,
largely due to overreduction or meisenheimer complex formation (HS-) and hydrolysis of a benzoquinone oxime intermediate. The economy of an aquous
solution and a cheap industrial reagent like NaS are just a logical choice, so not a lot of research would have been done if such a proven synthesis
exists.
The chemistry of ascorbic and dehydroascorbic at different pH levels is immensly complex, it indeed seems that ascorbic acid itself can form various
products when heated in aquous solutions, depending on pH and air exposure. This was why I was contemplating running the reaction at the lowest temp
possible for the reduction, namely 60 C.
"Degradation of Ascorbic Acid in Aqueous Solution
Jian-Ping Yuan, and Feng Chen*
Department of Botany, The University of Hong Kong, Pokfulam Road, Hong Kong
J. Agric. Food Chem., 1998, 46 (12), pp 5078–5082"
Considering the additional presence of Cu(II), Cu(I) being able to complex with some of these products and the formation of condensation products,
schiff bases and additional reducing agents from dehydroascorbic and decomposition products, it seems a small miracle picramic acid can be obtained in
this purity and yield.
Something I would still like to test are other transition metals (especialy iron) and whether the specificity of the reduction is largely a feature of
Cu(I) by using some other reducing agent. I can't see a direct reason though why Cu(I) would display such specificity. Anyway, if not, this would
also be further evidence the entire reaction may indeed be facilitated by the ligand coordination of ascorbic mediated by an insoluble
Cu(I)(ascorbic)picrate complex. Although far from proven at this point, it would be a sexy mechanism (Admittedly, I'm biased), the Cu(I) bringing
those reductive ascorbic hydrogens in perfect position for the reduction, almost like an enzyme would indeed.
https://en.wikipedia.org/wiki/L-ascorbate_oxidase
[Edited on 13-2-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
That was the impression I got from historical references. The description would indicate that picric acid alone even resists reduction in an acidic
system and is completely unreactive towards H2S until a base is added, whereupon reduction commences immediately.
| Quote: |
What would happen if a solution of picric acid in gAA or other solvent would be reduced with H2S, |
reportedly no reaction for H2S in absence of a base. With an equimolar amount of base available the reduction yield is quantitative.
| Quote: |
stannous chloride, zinc or iron powder e.g., never seen any references for this at least. |
These are
described to work with zinc and acetic acid, and iron powder with HCl, in references in the picramic acid from picric acid thread.
http://www.sciencemadness.org/talk/viewthread.php?tid=433&am...
http://www.sciencemadness.org/talk/viewthread.php?tid=433&am...
The reduction mixture scheme for using zinc dust for reduction of nitroguanidine to aminoguanidine is probably directly applicable to reduction of
picric acid to picramic acid.
| Quote: |
My uneducated thinking (without much reading) was that the impurities arising from the sulfide reduction are actually due to the high pH employed
during the reaction, largely due to overreduction or meisenheimer complex formation (HS-) and hydrolysis of a benzoquinone oxime intermediate. The
economy of an aquous solution and a cheap industrial reagent like NaS are just a logical choice, so not a lot of research would have been done if such
a proven synthesis exists.
The chemistry of ascorbic and dehydroascorbic at different pH levels is immensly complex, it indeed seems that ascorbic acid itself can form various
products when heated in aquous solutions, depending on pH and air exposure. This was why I was contemplating running the reaction at the lowest temp
possible for the reduction, namely 60 C. |
It isn't that sensitive to heat. Probably 85 C is a safe upper limit.
| Quote: |
"Degradation of Ascorbic Acid in Aqueous Solution
Jian-Ping Yuan, and Feng Chen*
Department of Botany, The University of Hong Kong, Pokfulam Road, Hong Kong
J. Agric. Food Chem., 1998, 46 (12), pp 5078–5082"
Considering the additional presence of Cu(II), Cu(I) being able to complex with some of these products and the formation of condensation products,
schiff bases and additional reducing agents from dehydroascorbic and decomposition products, it seems a small miracle picramic acid can be obtained in
this purity and yield. |
Hmmmm how about glycine ? Take a shot in the dark...and listen...maybe then a scream is heard in the distance
Magnesium is even more sure to make somebody holler.
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
| Quote: |
Something I would still like to test are other transition metals (especialy iron) and whether the specificity of the reduction is largely a feature of
Cu(I) by using some other reducing agent. I can't see a direct reason though why Cu(I) would display such specificity. Anyway, if not, this would
also be further evidence the entire reaction may indeed be facilitated by the ligand coordination of ascorbic mediated by an insoluble
Cu(I)(ascorbic)picrate complex. Although far from proven at this point, it would be a sexy mechanism (Admittedly, I'm biased), the Cu(I) bringing
those reductive ascorbic hydrogens in perfect position for the reduction, almost like an enzyme would indeed.
https://en.wikipedia.org/wiki/L-ascorbate_oxidase
[Edited on 13-2-2018 by nitro-genes] |
Tin and hydrochloric acid will reduce picric acid all the way to triaminophenol, but in alkaline condition, the reduction is limited to one nitro
group and picramic acid is produced.
https://books.google.com/books?id=pZDPAAAAMAAJ&pg=PA143&...
Previously in my post above on 2-5-2018 is an attached file about diglycine picrate
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
On page 5 of that article is described a hot solution of 20 ml H2O
in which are dissolved 3 grams of glycine plus 9.2 grams picric acid. That would be a solution that is 40% w/w picric acid which is an extraordinary
increase in solubility for picric acid. It is likely that a substantial amount of magnesium oxide or magnesium carbonate could be added to form some
magnesium picrate which is also highly soluble. My hypothesis is that due to the low solubility of the picramic acid and the higher acidity of the
picric acid that free picramic acid may precipitate directly from that acidic reduction mixture as fast as it forms from a reduction of the magnesium
picrate by free ascorbic acid added gradually as a hot nearly saturated solution. If the magnesium preferentially associates with the more acidic
picrate, being displaced from the picramate, then it will not require much of the magnesium value to function in a way that is similar to a catalyst.
As each magnesium picrate is reduced to picramate, the local acidity of free picric acid may associate with and displace magnesium from the nascent
magnesium picramate, precipitating free picramic acid, as the reduction proceeds. This magnesium would also protect the picric acid against reduction
of more than one of its nitro groups. This is simply my idea of what could and may occur, and I have no reference because i have never seen this
reported, it is purely a hypothetical and experimental suggestion which seems a reasonable intuitive approach. Making the addition of some of the
magnesium value in the form of magnesium ascorbate at some percentage perhaps a third of the ascorbic acid solution containing the magnesium ascorbate
may be a more refined method.
Confidence is high that some variation on this scheme could be optimized for reduction of picric acid to picramic acid. Variations might include
concurrent streams of separate additions of reactants into the reduction reaction mixture to maintain a controlled reaction conditions window at
optimum pH and proportions. An HCl reactant could be used if needed to manage pH by displacement of accumulating "spent" Magnesium sequestered as the
chloride if that is required. The MgCl2 would be an inert spectator split away from any Mg picramate converted to free picramic acid if that
disassociation of magnesium picramate is not a result occurring automatically in the reaction mixture due to the acidity of unreduced picric acid.
Precursor reactants that could be useful are already OTC nutritional supplement products. Glycine, magnesium biglycinate, magnesium ascorbate,
ascorbic acid, ect. are easily available. Epsom salt and sodium bicarbonate are other sources for precipitated magnesium carbonate that could supply
the magnesium value, as could be supplied in the alternative a similar magnesium hydroxide.
[Edited on 2/14/2018 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
in the other thread you asked a question about the Piria reaction
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
Bisulfite is one of the reducing agents I had predicted would likely produce a picramate from a picrate. It would probably work even better with an
added mole equivalent of NaOH to convert the bisulfite to the normal sulfite.
Picramic acid as sodium picramate is what I think will result without any indtroduction of a sulfonic meta to the amino in the case of a picrate due
to the meta position with regards to the amino, already occupied by a nitro, plus in alkaline condition the sulfonic acid would not be expected to
attach to the ring anyway. The reducing activity of the sulfite would still be active selectively towards the one nitro group at (2) ortho. Thus
should be formed sodium picramate from the reduction of sodium picrate by sodium bisulfite + NaOH which is equivalent to normal sodium sulfite.
[Edited on 2/15/2018 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
FR2106904 French patent DDNP
Diazodinitrophenol - as a free running crystalline powder from a nitric acid diazotisation
Abstract:
Diazoltisation of picramic acid to produce the explosive is effected in nitric acid in the absence of hydrochloric or sulphuric acids or their salts
and pref with as low a sodium content as possible. Conventional products tend to adhere to surfaces.
A translation of the attached patent FR2106904 may be useful
Attachment: FR2106904A5 Diazodinitrophenol - as a free running crystalline powder from a nitric acid diazotisation.pdf (196kB)
This file has been downloaded 635 times
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks for the patent and all the other information, not much time is left in the day and most of what is left is spent in "the lab". The plan is to
eventually start reading as well.
Did some small scale experiments again:
1. The initial light green precipitate formed from the copper/ascorbic reduction seems to contain unreacted picrate and Cu(I) ions
2. Although the initial complex (imediately after it formed) contains hardly any picramic, keeping at 40-50 seems to produce picramic acid eventualy,
while no gas formation is observed, Either this is occuring at a rate too slow to be noticed, or the gas formation is not a requisite for reduction
at all, in which case a longer heating period at 50 deg C could produce a purer product in higher yield. (To be tested at slightly larger scale)
3. After the heating period, the yellow-green precipitate also seems to contain Cu(I), probably a complexed or uncomplexed Cu(I)picramate. (After HCl
treatment and adding NaOH, a yellow precipitate of Cu(I) oxide is formed, adding HCl again and oxidizing the Cu(I) with nitrous acid and adding NaOH
again, greenish blue Cu(II)hydroxide is formed, which upon heating forms black Cu(II)oxide). This explains why at least 2 molar equivalents of Cu(II)
salts relative to picric are needed, and this would indeed mean that a larger amount of ascorbic acid is needed (>3.5 eqvts), like Rosco suggested.
4. A strange observation, somehow, the dark brown filtrate after the reduction at 70 deg C, reacts with NaCl to produce a clear orange solution, while
no Cu(I)chloride is precipitated. This suggest all copper is sequestered in the precipitate and a slight excess may produce higher yields. Although
the 0.01 molar scale is perhaps too small to produce accurate yield measurements, the ratio of the putative copper picramate and picramic obtained
after HCl treatment suggests that not all the copper picramate truly is copper picramate. Perhaps a part of the picramic remains in the dilute HCl
solution, or maybe a complexed copper picramate is obtained, or another copper salt is present from decomposition products of the ascorbic, or
unreacted picrate is still present, not sure. Regarding the reaction with Cl-, would a dinitro benzoquinone or an oxime or something be able to react
with Cl-?
5. Cu(I) and Cu(II) chloride are very soluble in excess HCl, stirring the copper picramate obtained with 10% HCl at 20-60 deg C is enough to displace
all the copper. About 1 gram of the putative copper picramate was added to a beaker and with slight heating, 10% HCl was added in small increments.
The displacement can be followed by watching the edge of the fluid level in the beaker. As long as a milky greenish colour is observed it needs more
HCl. In total 1 gram copper picramate needed about 12.5 ml of 10% HCl. Amazingly, from an amorphous copper picramate, reasonably large and compact
crystals of picramic seem to be formed this way, that take up much less volume then picramic obtained from the boiling HCl-dilution method I posted
earlier. It collects as a dense layer of beautiful pom-grenade coloured crystals at the bottom of the beaker, which can be decanted to near dryness
after only few minutes of standing. If a high yield synthesis is realized, it might be enough to simply wash and decant the copper picramate a few
times and then add HCl to directly yield picramic without any filtering and drying steps.
6. Copper(I)picramate seems to be oxidized in the air on longer storing. ALthough this doesn't seem to affect the picramic obtained, it seemed
Cu(I)picramate needed less HCl to displace the copper than a longer stored and presumably oxidized Cu(II)picramate.
[Edited on 17-2-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Here are some more old references
Attachment: Pages from Journal_Chemical_Society_London pg429-430.pdf (205kB)
This file has been downloaded 572 times
Attachment: php1yxne4 (177kB)
This file has been downloaded 569 times
From what I am understanding from obscure early references that I have not obtained but are described by others, it would seem that it is not strictly
required that the reduction mixture be alkaline but only that it contain picrate. So the reaction mixture may be mildly acidic and buffered by the
picrate in a system that is acidic yet contains some picrate, and the reduction occurs on the picrate. References to picric acid being boiled with
iron or zinc where a ferrous picrate or a zinc picrate exists and limits the acidity as a buffered system, present a reaction pH mildly acidic, yet
the iron or zinc picrate salt contained is susceptible to reduction to picramic acid or a picramate.
As an example, there is reported that iron powder and NaCl being boiled with picric acid will produce picramic acid. The particulars are not
described, but evidently the reduction occurs via one ferrous picrate acting to reduce another ferrous picrate to ferrous picramate which would likely
be disassociated by the excess acididty of free picric acid, recycling the ferrous value as the reduction continues, precipitating free picramic acid.
If the reduction would proceed similarly using ascorbic acid and a magnesium picrate, the advantage would be no insoluble or low soluble metal ions
to complicate isolation or work against the purity of the desired end product. There is no prior art reported for the ascorbic acid / magnesium
picrate scheme with or without glycine.
The activity of copper as the cuprous acetate or cuprous chloride is reported in the early literature, so there is prior art there even though the
detailed report is not found. Evidently the same complication of a copper picramate precipitate was observed which quenched enthusiasm for the
reduction scheme using copper.
The reduction using copper is interesting but the low solubility of whatever copper picramate compound or complex is causing precipitation and
sequestration of the copper during the reduction works against the economics and practicality of copper to achieve an economically viable process
without applying some scheme for recovery and recycling of the spent copper byproduct.
An efficient economically viable reduction scheme is going to produce a quantitative or nearly quantitative yield using cheap materials for the
process.
[Edited on 2/18/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Did another experiment again:
2.30 g (0.010 moles) of picric was suspended in 75 ml of dH2O, brought to 60 deg C and the pH adjusted using 10% NaOH solution to a pH of 7.
2.75 g (0.011 moles) of copper(II)sulfate pentahydrate was added at once and stirred to dissolve
7.00 g (0.040 moles) of ascorbic acid were then added at once and allowed to stirr for 45 minutes at 70 C.
Copper(I)picramate filtered off, washed thorougly and dried. Then gradually 10% HCl was added at 40 deg C until no more greenish milky colour of the
supernatant was visible. This was allowed to cool down to 4 deg C for serval hours and then filtered off.
Yield of copper(I)picramate : 2.44 g (93.2%)
Yield of picramic (from HCl): 1.47 g (73.8%)
Also did an HCl treatment recovery experiment:
Dissolved 1.99 grams of picramic in 15 ml of boiling azeotropic HCl, then diluted this with water to 100 ml and left at 4 deg C for 16 hours. Recovery
of picramic after filtering washing and drying (as in the other experiments) was 1.83 grams (92%). Considering a small additional loss of the
copper(I)picramate during washing filtering and transfering to weighing containers etc and the 0.01 molar scale of the experiment in general, the true
yield of the reduction itself likely is already around 85-90%.
There are still many variations possible, e.g. lower temperatures (40-50 C) for longer times, gradual additions of ascorbic, running at slightly lower
pH, using other bases to neutralize the picric than NaOH, or using even larger excess of copper(II) and ascorbic, though yield already seems quite
good.
I'll give magnesium (like Rosco suggested) and iron a shot as well, already tried zinc on a very small scale, though this did not seem to form any
insoluble complex or accelerate the reduction like Cu(II) did.
[Edited on 19-2-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
There is a discrepancy in the conversion of the cuprous picramate to free picramic acid that should not be there, suggesting the cuprous picramate is
impure, possibly impure with some of the cupric salt or basic cupric salt. If that is what is occurring the following may help.
Suggest instead of NaOH as the base, substitute MgCO3 or MgO or Mg(OH)2 and adjust pH to about 3 pH, increase the amount of CuSO4-5H2O to 3 grams, and
the ascorbic acid to 8 grams.
Copper Acetate may be a better choice there than the sulfate.
Copper sulfate could be converted to the carbonate or basic carbonate and that is then easily converted to the acetate by acetic acid. There could
also probably be used glycine (aminoacetic acid) to obtain the glycinate if that is of interest.
Thinking it over, I believe the SO4 component due to its electrical charge could inhibit and work counter to the scheme of attempting to have a
distinctly acidic reduction mixture kept acidic by unreacted picric acid, yet controllably buffered by a soluble picrate intermediate of inherently
limited alkalinity like the Magnesium picrate would be, as a strategy for optimizing the activity of the ascorbic acid in the acidic pH range of 2 to
4. So I am pretty certain copper acetate or copper carbonate should be used to avoid the sulfate ion. Keeping out the stronger base and stronger acid
values would tend to limit the potential extremes of pH and make the desired reduction pH window easier to regulate and optimize for ascorbic acid.
There are at least 4 different reaction pathways possible to be occurring simultaneously both with regards to the reduction by ascorbic acid /
ascorbate and with regards to the precipitated picramate constitution being cuprous picramate or a mixture with other possible copper values. So there
is an algebra that applies where the net reaction is an algebraic sum of the different reactions that varies according to the extent to which the
different reactions each contribute to the total reduction.
[Edited on 2/20/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
I call it a day, lump-and-dump-easy-OTC-picramic...whoohoooo
Suggestions welcome
[Edit, new file uploaded, some mistakes corrected]
[Edited on 5-3-2018 by nitro-genes]
Attachment: Reduction of picric to picramic using copper sulfate and ascorbic acid - Copy.pdf (885kB)
This file has been downloaded 802 times
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Woow nitro-genes! This method certainly beats the pants of the sulphide methods, can't wait to give it a whirl!
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Some mistakes were present in the first version, uploaded a new version in the previous post.
@Boffis
Reprtedly, the sulfide methods do give higher yields, though this is an easy reaction indeed using OTC chemicals. Higher yields may be possible somehow, though optimizing such undescribed reactions
is very labour and material intensive, especially as an amateur. Would be interesting to hear if you have some additional ideas how to possibly
increase yield further. Maybe a buffer solution at a pH of 3 like rosco suggested and gradual additions of ascorbic would be an improvement. Any
suggestions?
|
|
|
| Pages:
1
..
26
27
28
29
30
..
33 |
|








