| Pages:
1
..
13
14
15
16
17
..
19 |
aga
Forum Drunkard

Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Aw crap. That means i got to do it right, all the way through.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Yeah, but it doesn't have rhyme!
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Memory lane: MrHomeScientist and my efforts at alpha-terpineol from turpentine: forgot just how much effort has already gone into this!
http://www.sciencemadness.org/talk/viewthread.php?tid=15171&...
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
Wouldn't dumping a grignard (MeMgI or EtMgBr perhaps) into menthone be a rather more straightforward approach to this? Sure, it won't produce
alpha-terpineol, but it will produce an 11-carbon (or more) tertiary alcohol with an estimated b.p. of ~220C (for the methyl derivative).
Menthol is fairly cheap and it's conversion to menthone is straightforward and high-yielding. http://www.orgsyn.org/demo.aspx?prep=CV1P0340
[Edited on 16-1-2016 by UC235]
|
|
|
HeYBrO
Hazard to Others
Posts: 289
Registered: 6-12-2013
Location: 'straya
Member Is Online
Mood:
|
|
Quote: Originally posted by blogfast25  | | Quote: Originally posted by aga |
Excellent ! Some actual data to work with ! Woohoo !
Nice find.
| Quote: Originally posted by Nicodem | | Try stirring a solution of pinenes or limonene in acetic acid in the presence of 1 mol% conc. sulfuric acid. A sulfonic acid such as MsOH and TsOH, or
sulfamic acid, would be preferable over sulfuric acid |
Not looked up what MsOH or TsOH are, however there does seem to be an unopened bottle of sulphamic acid on the shelf.
'acetic acid' ?
Does that mean glacial acetic acid or would vinegar do ?
The Brandy has almost all gone : it's heading the right way for a whole pile of TCA.
|
GAA = pure acetic acid. It's what he meant. Vinegar is only good for fish and chips.
TsOH: ortho-toluenesulphonic acid.
You are the Keith Floyd of amateur OC and the Keith Moon of casual alcoholism!  
[Edited on 12-1-2016 by blogfast25] |
Nitpik: TsOH is usually encountered as para-toluenesulphonic acid as the ortho won't form easily due to steric hinderance, so i pressume the "p-" is
left of for convenience.
UC, i have performed the oxidation of menthol to menthone with jones reagent and had good yields. The procedure is incredibly simple.
[Edited on 16-1-2016 by HeYBrO]
[Edited on 16-1-2016 by HeYBrO]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by UC235 | Wouldn't dumping a grignard (MeMgI or EtMgBr perhaps) into menthone be a rather more straightforward approach to this? Sure, it won't produce
alpha-terpineol, but it will produce an 11-carbon (or more) tertiary alcohol with an estimated b.p. of ~220C (for the methyl derivative).
Menthol is fairly cheap and it's conversion to menthone is straightforward and high-yielding. http://www.orgsyn.org/demo.aspx?prep=CV1P0340
[Edited on 16-1-2016 by UC235] |
I fear 11 (or more) would already be too long, as there's a balance between increased solubility of the K/Na/Mg t-alkoxides and steric hindrance to be
struck.
Similarly, I don't think Grignarding isopropylmyristate would yield anything all that useful.
One that could be worth looking at is 2-phenylpropan-2-ol: relatively easy:
benzene > bromobenzene > 2-phenylpropan-2-ol
[Edited on 16-1-2016 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
All too complex, especially for me - no bottle labelled 'Grignard' can be seen.
Doubt i could get the apparatus dry enough for a Grignard and i have no idea how they work anyway.
Rebooted the whole TCA thing by er, 'disposing' of the crappy ethanol and Properly distilling more (Vigreux, 1 C differential, done twice) plus making
fresh K3PO4 to make this batch of ethanol Actually dry.
Simultaneously triple recrystallising some agricultural KNO3 (+48% K2O) for the nitric step after chloral hydrate exists in the
shed.
Wondering about the N2O4 disposal/neutralisation, as plumes of orange/red gas are not generally welcomed.
Bash it into Water seems the obvious idea. Maybe iced ?
[Edited on 16-1-2016 by aga]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga | All too complex, especially for me - no bottle labelled 'Grignard' can be seen.
Doubt i could get the apparatus dry enough for a Grignard and i have no idea how they work anyway.
Wondering about the N2O4 disposal/neutralisation, as plumes of orange/red gas are not generally welcomed.
Bash it into Water seems the obvious idea. Maybe iced ?
|
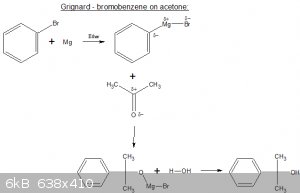
Step I: bromobenzene is reacted with an electropositive metal (here Mg) to form an organometallic bromide (aka 'Grignard reagent').
Step II: add acetone (in this case), an adduct is formed.
Step III: adduct is hydrolysed (usually with weak HCl), carbinol is formed. Separate as per usual.
Grignard reagents are very sensitive to water but I do believe some SMers are a bit paranoid about that.
**********
N2O4 disposal: yes, water or weak NaOH will absorb it very efficiently. BUT. Oxidations using HNO3 actually produce
NO, NOT NO2:
NO3<sup>-</sup> + 4 H<sup>+</sup> + 3 e<sup>-</sup> === > NO + 2 H2O
The NO2 you see is because of spontaneous oxidation of NO by air oxygen:
2 NO + O2 === > 2 NO2
So for the NaOH absorption scheme to work there must also be plenty of air oxygen present. Usually that is the case.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
A t-alcohol (carbinol) that hasn't been considered so far is:

1. De-esterify iso-amyl acetate ('banana oil', quite OTC) to get iso-amyl alcohol, aka 3-methylbutan-1-ol (there's a thread on that)
2. Chlorinate iso-amyl alcohol with HCl/anh. ZnCl2 to 1-chloro-3-methylbutane (iso-amyl chloride).
4. Grignard with Mg and acetone to 2,4-dimethylpentan-2-ol.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Maybe. Got to try the turps + TCA route first.
Had to calcine some limestone today as i'd run out of CaO needed to purify the phosphoric acid needed for the tripotassium phosphate needed to dry the
ethanol needed for chlorinating to chloral needed to make TCA needed to catalyse the turps to terpineol reaction.
Sorry it's taking so long 
Good news is that there's lots of nice clean KNO3 now for making WFNA for the nitration step.
Edit:
The K3PO4 didn't do diddly in drying ethanol as the phosphoric acid and the pot hydroxide were both Lepers (unclean !).
[Edited on 18-1-2016 by aga]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga |
Had to calcine some limestone today as i'd run out of CaO needed to purify the phosphoric acid needed for the tripotassium phosphate needed to dry the
ethanol needed for chlorinating to chloral needed to make TCA needed to catalyse the turps to terpineol reaction.
|
Hmmm... how do you calcine CaCO3 to CaO? I mean, the process has been used since Roman Times but it does require long
calcination times (up to 24 h at 1000 C, IIRW).
Also, if you're looking to dehydrate azeoptropic EtOH then CaO might be your thing: quicklime is indeed used in the production of bio-ethanol, if
water-free EtOH is needed.
Quicklime is of course quite easy to get: see building materials. It's also useful to make benzene with, from benzoic acid or sodium benzoate.
[Edited on 18-1-2016 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Stick lumps of limestone in a fire obviously !
Didn't know that the process took that long - this only ran for about an hour, max.
    
Top left to bottom right : furnace, raw rocks, rocks in crucible, view thru the lid whilst running, results + remains of copper.
The bit of copper pipe was in there to see if any part of the furnace hit 1000+ C.
The stones are cetainly whiter than before and quite crumbly.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga |
Stick lumps of limestone in a fire obviously !
Didn't know that the process took that long - this only ran for about an hour, max.
The stones are cetainly whiter than before and quite crumbly. |
Check progress by means of weight loss, which on completion should be 44 w%.
Also, freshly prepared CaO being a strong alkali it reacts quite violently with water. Apparently 'in Tempus Romanus' slaking the lime with water was
not a much envied job!
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
The Chloral synthesis isn't going well at all, hence no progress on the TCA synthesis.
After 3 sessions in the furnace the limestone had reduced to around 48% of the original weight, then used to dry the ethanol.
The result was still not 100% dry, however with no other drying agent on hand, chlorination was attempted, this time for 2 hours.
The outcome was exactly the same as before, with Zero white precipitate.
The Cl2 must be reacting with the ethanol/water, as the liquid heats up.
I suspect that either the 'chloral alcoholate' dissolves in the water or the reaction takes a different route altogether due to the water.
The only time a precipitate was seen was with anhydrous ethanol prepared with oven-dried tripotassium phosphate, so 120g of that has been made today,
this time starting with stoichimetry and calculations instead of a beaker full of hope.
Remarkably the yield is almost exactly the same as the calculated theoretical, so should work as expected.
Unfortunately all the booze seems to have gone, so Shopping & Distilling tomorrow.
[Edited on 24-1-2016 by aga]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Did you mean weight loss was about 48 %?
How much quicklime per volume of EtOH?
Did you crush the quicklime to powder?
What was the contact time?
How did you test EtOH dryness?
<hr>
I also think it might be useful to check the calibration of your refractometer.
Combining data from this page:
http://uk.mt.com/dam/Analytical/Density/DE-PDF/BRIX-Sugar_De...
and this page:
http://www.refractometer.pl/refraction-datasheet-ethanol
I deduce that a solution of 17.5 degrees Brix should give a reading of 44 % ABV on your refractometer.
17.5 degrees Brix = 17.5 g sucrose (white sugar) per 100 ml of pure water.
[Edited on 24-1-2016 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
| Quote: Originally posted by blogfast25 | Did you mean weight loss was about 48 %?
How much quicklime per volume of EtOH?
Did you crush the quicklime to powder?
What was the contact time?
How did you test EtOH dryness?
<hr>
I also think it might be useful to check the calibration of your refractometer.
...
I deduce that a solution of 17.5 degrees Brix should give a reading of 44 % ABV on your refractometer. |
In the end there was just under 95g of useable CaO from the original 197g of limestone.
Accurate weights are pretty much impossible when the substance is in a charcoal furnace with a makeshift lid on the crucible as bits of charcoal get
in, some stone becomes powder, and all have to be discarded.
All of the useable (alleged) CaO was crushed and added to 200ml of ethanol, then left to stand for around 30 mins.
Dryness was tested with KMnO4 as a quick way to show if the water content was significant.
Had no purple colour appeared (which it did) i'd have diluted the ethanol 1:1 and used the ethanol refractomer rather than the other
sucrose refractomer next to it.
With luck there will be dry ethanol after distilling some crude tomorrow, then applying the tripot.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
30 minutes seems like a short time to me. Overnight should work well, I think.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
The dehydrating effect with recently oven-dried tripotassium phosphate is almost instant, also complete (as far as i can tell) so preferred.
Also preferred due to the fact that a white precipitate was only seen when chlorinating tripot-dried ethanol.
Perhaps water in the Cl2 stream is why that experiment seemed to halt after a few minutes.
Definitely must add a stage to dry the Cl2 on the next attempt.
Any clues as to what Liquid product ethanol + Cl2 + H2O makes instead of a Solid chlorinated ethanol thingy ?
The products of the failed chlorination attempts smell nice, but not like 'musty hay' (thankfully) and are out in a field evaporating away.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga |
Any clues as to what Liquid product ethanol + Cl2 + H2O makes instead of a Solid chlorinated ethanol thingy ?
|
Maybe small amounts of water increases the chloral hydrate's solubility by a lot?
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
As far as the meagre information goes from the paper, you need to get the Solid 'chloral alcoholate' first, then add conc sulphuric acid to that to
get the Liquid 'chloral'.
After that, separate, then add equimolar water to get the Chloral Hydrate.
All unexplored territory to me, so i really have no idea.
Chemistry is great fun.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Here's an idea:
If you don't get any crystals, why not try and distil off some of the ethanol? The BPs of azeo EtOH and chloral hydrate are quite different.
The chloral hydrate is very soluble in ethanol, so by distilling some of it off you may reach the solubility limit of the chloral hydrate, then it
should crystallise on cooling.
I don't think water impedes the actual synthesis: choral (not hydrate) can also be made from acetaldehyde (the aldehyde of ethanol), Cl2 an
HCl solution. Water doesn't seem to be the problem.
[Edited on 25-1-2016 by blogfast25]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
If you continue to have issues with the synthesis, I would recommend an alternative synthesis that circumvents the need for dry ethanol and chlorine
gas. It involves reacting trichloroethylene with a source of hypochlorous acid, such as an acidified hypochlorite, or TCCA/NADCCA which is more
convenient. IIRC the reaction takes place at STP and takes a few hours. There is a thread on SM somewhere, as well as a US patent detailing the
process.
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Here is thread 1 : http://www.sciencemadness.org/talk/viewthread.php?tid=2535&a...
thread 2 : http://www.sciencemadness.org/talk/viewthread.php?tid=62148
and patent : https://www.google.com/patents/US2759978
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Thanks, gdflp!
From the first thread:
| Quote: Originally posted by erich_zurich | I talked about synthesis of chloral hydrate from trichloroethylene and hypochlorous acid. I also tried a improved synthesis of chloral hydrate from
ethanol, chlorine gas and by utilizing UV light to initiate the free radical chlorination reaction, I will describe both procedures.
Method #2 MY FAVORITE! SEVERAL TIMES FASTER THAN THE ORIGINAL CHLORINATION METHOD!
100 ml of absolute ethyl alcohol is placed in a ice cooled flask with a black light 10cm away from the flask irradiating it with Ultraviolet light. to
the flask a bubbler is added to which a current of dry chlorine gas is passed by maintaining the temperature below 10° C. The chlorine is quickly
absorbed, and, after a short time, the reaction flask is connected with a reflux condenser. By gently warming of the liquid to 60° C the saturation
of the solution is continued with chlorine gas until it is fully absorbed. The chlorination reaction is complete when the solution reaches density of
1.4 g/ml, then the liquid is gently boiled for a short time and allowed to cool after which it is cautiously mixed with an equal volume of
concentrated sulfuric acid. During this addition hydrochloric acid and ethyl chloride are evolved. Then the reaction mixture is distilled and the
distillate is neutralized with calcium oxide or carbonate and redistilled. Further purification is performed by fractionation. Remaining ethyl
chloride with hydrogen chloride distills off first. Between 70° C and 90° C ethyl alcohol passes over, and from 90° C chloral starts to distill.
Chloral is a colorless mobile liquid, which boils at 94.5° C. When mixed with about one-fifth of its weight of water, the mixture slowly solidifies
to a crystalline mass of chloral hydrate:
|
That sounds like a modified version of what aga is currently attempting.
Then the chloral (trichloroacetaldehyde) is oxidised with HNO3 to trichloroacetic acid.
[Edited on 25-1-2016 by blogfast25]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
| Quote: Originally posted by blogfast25 |
Thanks, gdflp!
From the first thread:
That sounds like a modified version of what aga is currently attempting.
Then the chloral (trichloroacetaldehyde) is oxidised with HNO3 to trichloroacetic acid.
|
Indeed, but I was talking about the first two in that thread. They're much more robust IMO, especially if aga is having issues with the ethanol
chlorination.
|
|
|
| Pages:
1
..
13
14
15
16
17
..
19 |








