| Pages:
1
2 |
Boffis
International Hazard

Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Nice work nitro-genes! 17g is a pretty good yield for such a brutal method and almost exactly the reported yield. Did you keep the brown stuff? my
understanding from the paper is that more picramide and some cyanuric acid can be recovered from it.
Somewhere amongst my papers I have one on the action of hydrogen sulphide on picramide to produce 3,5-dinitro-1,2-diaminobenzene
(3,5-dinitro-o-phenylenediamine). I´ll dig it out but its not an obvious title as a quick searchy on my HD failed to find it.
Found it! it´s a whole-volume pdf so I´ll have to do a bit of pruning but here is the ref.
Norton & Elliott, Berichte v11, p327 (1878)
I´ve posted this ref before in the triazole thread so I may have been posted under the references wanted section already.
[Edited on 12-5-2015 by Boffis]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The brown stuff is partly a condensation product of the cyanuric acid and formed picramide indeed, I thought about acidifying the brown suspension
using HCl and boiling it for some time to hydrolyse, possibly resulting in a slightly higher yield. Addition of a dipolar aprotic solvent would be a
much safer synthesis, the use of sulfolane for the TNP-TNA conversion at high temperatures has been described and patented, can anyone think of
another possible OTC solvent, maybe propylenecarbonate?
https://e-reports-ext.llnl.gov/pdf/332580.pdf
US7057072
For the reduction, alternatively, ascorbic acid might be useful as well. Supposedly, dinitro and mononitro anilines cannot be reduced by ascorbic
acid, which could mean that ascorbic acid might be able to robustly reduce a single nitro group for TNA. When I boiled a very small amount of TNA/NaOH
with NaAscorbate, a brown powder came out of solution, which would be the most likely colour for the diamino, dinitrobenzene. Will try this on larger
scale soon.
[Edited on 12-5-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Quote: Originally posted by nitro-genes  | The synthesis of trinitroaniline from picric acid and urea works perfectly as described. In a beaker with teflonized (gas tape) lid, 23 grams of
finely powdered recrystallized TNP was intimately mixed with 18 grams of finely powdered urea. This was (outside in the shed) heated for 24 hours at
180 deg C in a small electric oven. An extra oven temperature indicated that the temperature remained between 170 and 178 degrees C at all times. The
mix melted producing an orange liquid, that gradually became a distinct dark red. White fumes were emitted during the reaction, consisting most likely
of NH3 and CO2, probably recombining to ammonium carbonate in the air, giving the white fumes.
After 24 hours at 175 and cooling, 150 ml of water was added. It was a PITA to get it all to form a suspension, forming a glassy subtance, that needs
considerable effort to break up under water. The suspension was filtered and after drying of the resulting brown powder, was dissolved in 150 ml
acetone at 40 degrees. Brown insolubile stuff remained, while an orange solution of TNA was obtained. To this was added very slowly while stirring on
the hotplate, 150 ml water and then boiled until no smell of acetone could be noticed. This gave 17 grams TNA of a orange/yellow colour, melting point
around 180-190 ish (hotmplate, so not very acurate). :-)
I have a hunch for a OTC procedure for the selective reduction of one ortho nitro group, will post results in a few days. :-)
EDIT: This procedure is dangerous enough as it is, and only to be performed outside and with recrystallized TNP. Do not use a metal lid for the
reaction, volatilization of the picric acid was evident, creating a yellow film covering the entire reaction vessel. Contamination with metal picrates
can possibly lead to detonation of the entire mix!!! Also make sure that both the TNP and urea are finely powdered and mixed PRIOR to heating/melting.
[Edited on 11-5-2015 by nitro-genes] |
Interesting. I must wonder what would be the result if the Spencer and Wright synthesis was applied to styphnic acid instead of picric acid. Using
the same proportion for urea would one of the hydroxyls of the styphnic acid remain intact, possibly to combine with byproduct ammonia?
Also there would seem to be safety concerns about this reaction for reason of the elevated temperatures and the nature of the nitro compounds being
heated. So I must wonder also if danger lurks which is an unquantified "variable" 
[Edited on 12-5-2015 by Rosco Bodine]
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@nitro-genes. I have seen that patent before but had forgotten about it. However, it has given me an idea.
Instead of sulfolane how about refluxing DMSO, this compound can be purchased easily on line and is fairly cheap and boils at 189 C only slightly
above the required reaction temperature. Another possible solvent is N-methylpyrrolidone with a slightly higher boiling point (202 C)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
For a solvent moderating the reaction even something like kerosene or other middle paraffin petroleum could work, but will possibly slow down the
already slow reaction. Softening the solidified chunk from the unmodified reaction might be facilitated by hot xylene. If it (hopefully) forms a hot
syrup it could be streamed into boiling hot vigorously stirred H2O and a lot of the xylene will steam distill away leaving the particles in
suspension for later filtering and redissolving in acetone.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I think at least one of the reactant should be soluble in the solvent /dilutant. Ethylene glycol may also work and I have both n-heptanol (Bp 175 C)
and n-octanol (Bp 195 C) too.
Alternatively why not just use more urea as both a reaction and flux/dilutant?
[Edited on 12-5-2015 by Boffis]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Forming urea picrate first as an intermediate could also be useful.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
IMO, from a practical standpoint, regarding isolation of the TNA, the solvent needs to be soluble in (hot) water or acetone/water mixtures to some
extent. The water extraction to remove the excess urea after heating is already pretty messy, imagine a sticky viscous hydrophobic oil thrown into the
mix. :-) Might still work, if careful recrystallization is performed after the acetone extraction, leaving the solvent mostly in solution, but this
would need a lot of work probably to optimize. Moreover, if the reactant(s) are not soluble in the melt/solution then phase seperation will occur and
no reaction.
DMSO/propylene carbonate would probably a better option, though DMSO would have been such a logical choice, that it seems strange sulfolane is used in
the patent. I was thinking... some plasticizers are pretty water soluble and largely aprotic, triethyl citrate for example (used as food additive)
although I'm not sure if they can stand the reactants and conditions applied, without transesterification occuring with the TNA.
A very interesting candidate IMO: Dot4 brake fluid consists mostly of butoxy triglycol, although pretty much water insoluble. Dot 3 brakefluid is much
more water soluble IIRC, this is maybe the the best completely OTC solvent on my list, although it is still protic and will probably condense with
the urea. :-)
Adding more urea probably won't work, according to the Spencer paper, it leads to more cyanuric acid and more of the acetone insoluble brown stuff.
However...
From patent --> US3785796
"However, on searching the literature, it became obvious from an article in the Russian Journal of Applied Chemistry, vol. 40, 9, p. 1989 (1967) and
from the work done by Meiser, supra, that ammonium sulfate was slightly soluble in urea. The Russian article stated that a eutectic point existed at
121.5° C. and at this temperature, a mixture of 9 percent ammonium sulfate and 91 percent urea was liquid. "
Without a suitable OTC solvent, it might still be interesting to examine the outcome for some ammonium salts/urea eutectics, that may lead to less
formation of cyanuric acid, condensing with the TNA, lowering yield. This is probably the reason why ammonium salts are used in the patent.
@ Rosco, the idea of performing the reaction with TNR instead of TNP is an interesting idea! Does TNR have similar decomposition temperature compared
to TNP? If I'm able to find a suitable solvent, I might give it a shot...
[Edited on 12-5-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Checking further I don't think styphnic acid will work the same as picric acid heated with urea because the ammonium styphnate byproduct detonates on
heating.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Hmm, detonation of reactants is never a good thing...
Offtopic These KC300 nitrile gloves can easily rupture, I didn't notice but one fingernail was stained bright yellow from TNP solution. Who would have
thought one of these could be so convenient, I hope she didn't notice.
https://www.google.nl/search?q=nail+file&source=lnms&...
[Edited on 13-5-2015 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Had some dot 4 brakefluid lying around and diammonium phosphate (yeast nutrient salt for wine making, called DAP). Did some testing, looking at
stability and solubility without TNP added. Unfortunately, brakefluid does not dissolve DAP. When heated for some time with urea, a solution was
obtained, that after cooling solidified to one crystalline mass. TNA heated in BF does not dissolve, but is recoverable by adding hot water and
filtering. I'm out of TNP, so couldnt test solubility.
DAP gives among the best yields of TNA (87-94%) in sulfolane conversions and is easily and purely obtained as yeast nutrient base or fertilizers. All
we need is an OTC, polar aprotic solvent. I could find DMSO as health product online, but it was like 40 dollars for 250 ml.
[Edited on 14-5-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by nitro-genes | Ok, nitration went much smoother than anticipated. I did't use HNO3, I was out, and didn't want to distill something if nitrate salts could be used
instead. :-)
7.7 grams of absolutely anhydrous ammonium nitrate was dissolved in 20 ml 98% sulfuric acid at room temperature, this was put at -20., resulting a a
clear syrupy liquid. In a separate beaker, 60 ml of 98% sulfuric acid was poured and also put at -20. The next day, 9.4 grams of acetaminophen was
dissolved in the 60 ml of sulfuric acid (while on salted ice bath), only very slight exotherm occured, so addition of the AN/SA was directly done
after. Using a pipet the AN/SA was slowly allowed to drip in the SA/Acetaminophen solution, temperature did not went over 0 degrees at all times,
addition was done over the course of about 15 minutes, almost no smell of NOx was noticed, but very faint smell of acetic acid was. An interesting
note is that the solution went instantly from sort of orange to almost red with the addition of the last drops of AN/SA, which seems a good indication
for completion of the reaction. The mixture was poured in 300 ml of icecold water upon which a orange-yellow solid precipitated. This was washed with
another 300 ml of icecold water and collected to dry. It has low water solubility, stains like picric acid and sodium bicarbonate produced a bright
red solution. Looking at the solubility of the potassium salt right now, as it is in the fridge.
Some additional notes, exotherm is really manageable, salted icebath doesn't seem obligatory, crushed ice should work fine with maybe slightly longer
addition time. For the next synth I will try to minimize the amount of 98% SA needed.
The nitration also produced a small amount of sticky, chewing gum like stuff, probably left overs from the binder/sugars in the acetaminophen tablets.
Either that or polyanilline like derivatives may have formed during the reaction. It's probably better to either start with the pure stuff or do
proper recrystallization from ethanol first. (I simply evaporated everything at low temp).
Question remains which isomer we have produced, though the nitration itself seems to work. Also wonder how sensitive this stuff is to
hydrolysis/oxidation by air, with some of the entrapped acid remaining from the synthesis. Bicarbonate neutralization to produce the sodium salt may
be a better option, but how best to proceed for the hydrolysis/hypochlorite oxidation?
[Edited on 30-4-2015 by nitro-genes] |
In the DDNP thread there was some consideration of the nitration of paracetmol / acetaminophen as a route to isopicramic acid, and/or its acetyl
derivative and this was never completely sorted out from the literature references. But this may be what is your product. Your acetaminophen
nitration product may be isopicramic acid directly or after deacetylation, isopicramic acid, which may be diazotized to form iso-DDNP.
Depending upon conditions I think it is possible the nitration product could be a mixture of isopicramic acid and as an impurity the acetyl derivative
of isopicramic acid. Your noting some odor of acetic acid would indicate at least a partial deacetylation, and to what extent is unknown. If
deacetylation was complete then your product would be isopicramic acid. To whatever extent the deacetylation did not complete, the product would be a
mixture of the deacetylated and the acetylated forms.
In either case, your product is a precursor for diazotization to iso-DDNP. The interesting, intriguing possibility about iso-DDNP is that it may be a
better initiator than the better known DDNP. Also possible, iso-DDNP is more easily made from a precursor gotten directly from nitration which
requires no sulfide reduction of a trinitrophenol as is done to produce the ordinary picramic acid, since isopicramic acid can be gotten directly from
a mild reaction condition dinitration of acetaminophen, preserving the amino group already present on the ring. If iso-DDNP is a better initiator
produced by fewer steps from a more easily nitrated precursor, that could make iso-DDNP more desirable and economical than the more commonly known
DDNP. Potentially, iso-DDNP could prove to be superior and more easily and cheaply made than the better known DDNP.
See this post in the DDNP thread.
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
[Edited on 17-5-2015 by Rosco Bodine]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Oxydative Nitration of acetaminophen may also produce nitranilic acid via paraquinone.
HO-C6H4-NH2 -oxydation-> O=C(CH=CH)2C=NH
O=C(CH=CH)2C=NH + H2O -acid-> O=C(CH=CH)2C=O + NH4(+)
O=C(CH=CH)2C=O -HNO3-> O=C(CNO2=CNO2)2C=O
Ortho-dinitro suffers from nitro-nitrite switch so
O=C(CNO2=CONO)2C=O + 2 H2O --> O=C(CNO2=COH)2C=O + 2 HONO (H2O + NO + NO2)
O=C(CNO2=COH)2C=O is nitranilic acid but the sequence is
cyclo(-C(NO2)=C(OH)-CO-C(NO2)=C(OH)-CO-) so one side of the quinone is (-C(NO2)=C(OH)-) and the other is (-C(OH)=C(NO2))...
All pairs of groups are para to each other 2,5-dihydroxy-3,6-dinitro-para-quinone...
The two ketonic groups, the two nitro groups and the two hydroxy groups.
Note that by virtue of proton jump and resonance between enol-keton form: 2,5-dihydroxy-3,6-dinitro-para-quinone is also
4,5-dihydroxy-3,6-dinitro-ortho-quinone
Not counting with the nitro-nitronic resonance that allows the protons to jump from each group to the other one.
So any corner of the hexaring may hold a proton with a given resonance conformation.
Really a beautifull molecule.
[Edited on 17-5-2015 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Isopicramic acid from paracetamol seems an interesting possibility, the synthesis of N-methylated isopicramic acid is described in US3641154 and
supposedly gives good yields. Here they use N-methyl or N-isoproppyl derivatives for the nitration, so the question is if the N-acetyl group from
acetaminophen is protective enough to survive the nitration. If not, oxidation like philou pointed out might become more favorable. Thanks for working
that out, I was curious how the amine group would be replaced by the keto-enol group. Would the use of ammonium nitrate for the nitration be
protective, since excess NH4+ (although AN also partly dehydrates to nitramine in SA IIRC) would be present during the reaction? slwoing down the
O=C(CH=CH)2C=NH + H2O -acid-> O=C(CH=CH)2C=O + NH4(+) step? I used AN instead of NA or NaO3 since AN/SA generally produces very little NOx, which
would be the likely main oxidizing species in the mix, leading to paraquinones. (True?)
Another PITA of this synthesis I can see is the necessity to neutralize most of the mixed acids (ph 4)in order to precipitate the product. Not only
would it use a large amount of NaOH, but will lead to a huge exotherm in which most of the product is likely oxidized in the partly diluted acids. A
more sophisticated way to isolate the product may be beneficial, although perhaps the N-acetyl isopicramic acid is less soluble in the mixed acids
than the N-methyl and isopropyl products described in the patent. The (likely) mono nitro variant synthesized earlier did precipitate pretty well...
Speaking of an OTC route to DDNP:
Just for fun I thought of maybe another feasible, alternative route to picramic acid. (Acetyl)Salicylic acid --> 98% H20SO4, 110 deg. C for 45
minutes --> p-sulfosalicylic acid --> dilution to 25-50% H2SO4 --> 1/3 mol TCCA 40-75 deg. C --> 3-chloro sulfosalicylic acid -->
saturated NaCl and chilling, filtering --> nitration to 2-chloro 4,6 dinitrophenol --> ammonia --> picramic acid. 
1. http://repository.ias.ac.in/78824/1/78824.pdf
2. http://www.sciencemadness.org/talk/viewthread.php?tid=24147
Anyway, kind of busy right now, though I have some purified acetaminophen left so will give it a try soon 
[Edited on 18-5-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The nitration conditions of US3641154 are applicable.
It was GB24409 that described a nitration of paracetamol to produce the 2,6-dinitro, 4-acetylaminophenol which is the acetyl derivative of isopicramic
acid. In the patent this was an intermediate intended to be reduced to provide an amino derivative which was the interest of the patent. For our
purpose the dinitration product of paracetamol is de-acetylated by treatment with hydrochloric acid, which results in isopicramic acid. In the
alternative heating the dinitrated paracetamol with dilute sulfuric acid should likewise accomplish the de-acetylation under milder conditions than
hydrolysis using HCl and give isopicramic acid.
The analogous benzoylated compound being de-benzoylated by HCl to isopicramic acid was reportedly heated with HCl at 130C for 12 hours, which would be
autoclave conditions. So the method of de-acetylation by heating with dilute H2SO4 would seem to be more convenient.
See page 1204 (journal page number) of the journal article
by Meldola and Stephens attached. There is described an analogous de-acetylation using dilute H2SO4, saying the acetyl compound was "readily
hydrolyzed" by heating with dilute H2SO4. With no high temperature like 130C being stated as for HCl, this description would suggest ordinary
conditions for the de-acetylation by H2SO4.
A more complete description of the hydrolysis accomplished debenzoylation using H2SO4 is described in the Chemical World article page 327 by Meldola,
Hale, and Thompson. It seems probable the same procedure will likewise accomplish the de-acetylation in the same manner as de-benzoylation.
The isopicramic acid is then diazotized, to provide iso-DDNP
4-diazo-2,6-dinitrophenol.
Attachment: Berichte der deutschen chemischen Gesellschaft, Volume 38, Issue 2 (p 1593-1599).pdf (348kB)
This file has been downloaded 667 times
Attachment: Pages from Journal_of_the_Chemical_Society pg308.pdf (434kB)
This file has been downloaded 570 times
Attachment: Pages from Journal_Chemical_Society_London pg1203.pdf (380kB)
This file has been downloaded 495 times
Attachment: Pages from The_Chemical_world pg327.pdf (247kB)
This file has been downloaded 578 times
[Edited on 19-5-2015 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by APO | | In the first link I referenced [1], it links to a patent that specifically names the compound received by mixed acid nitration using HNO3/H2SO4 as
"3-nitro-4-substituted aminophenol", which I called 4-acetamino-3-nitrophenol. Meaning that for that particular process, among others, the nitro group
is usually added adjacent to the acetamino group. Most of what I have seen shows the compound resulting from nitration or nitrite addition as having
the nitro group in the meta (3) position, however I have also seen some references as having it in the ortho (2) position. All I can really gather
from this mix of data is that you can pretty much pick whatever isomer you want, however, what you get is very dependent on reagents used and reaction
conditions. |
Precisely. It seems quite nuanced according to conditions and perhaps order of addition and the ratio of reactants what is gotten as a result for the
first entering nitro group, and for second and third also. The course of synthesis can be finessed and steered when the factors which govern are
identified. It is very interesting and I have been straining my brain looking at this subject for more than a year, trying to sort it out from the
earliest references, including a Reverdin and Dresel article requested today which should provide original information about the
4-acetamino-3-nitrophenol, mp. ect.
Attached are 2 Reverdin and Dresel articles
On journal page 440 of the Archives des sciences physiques et naturelles by Reverdin and Dresel are several melting points stated for the m-nitro-p-aminophenol. I am still reading to find the acetyl derivative / precursor mp
which is 4-acetamino-3-nitrophenol, on page 442. The Chinese patent has later reported 162C mp for the acetyl derivative.
Japanese pharmacists have reported in 1989 the mp for 4-acetamino-3-nitrophenol as 139C
See attached file: A NEW NITRATION PRODUCT, 3-NITRO-4-ACETAMIDOPHENOL, OBTAINED FROM ACETAMINOPHEN WITH NITROUS ACID
Attachment: Reverdin and Dresel, Archives des sciences physiques et naturelles, 1904, 18, 433 to 444.pdf (392kB)
This file has been downloaded 522 times
Attachment: Mononitroderivate des p - Aminophenols. Mittheilnngen.-reverdin 1904.pdf (245kB)
This file has been downloaded 617 times
Attachment: A NEW NITRATION PRODUCT, 3-NITRO-4-ACETAMIDOPHENOL, OBTAINED FROM ACETAMINOPHEN WITH NITROUS ACID.pdf (178kB)
This file has been downloaded 573 times
[Edited on 6/15/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Many dinitrobenzofuroxans have been described (CL14,CL17 etc), although I've never seen a dihydroxy dinitro benzofuroxan. Wonder how stable it would
be, if it exists at all, seems it can form a very stable resonance structure.
Considering 1,4 benzoquinone and hydroxylamine are pretty OTC, how about this for a synthesis. All steps up to the nitration of
1-Chloro-3,5-dihydroxybenzene are desribed in literature and give reasonable to good yields.
The second reaction is interesting, never seen this type of rearangement/addition reaction type for a nitroso compound before, supposedly, it gives
high yields as well (87%). Is it possible o-nitrosophenol (o-benzoquinone monooxime) would also result in a 1-chloro-3,5 dimethoxyaniline derivative
from the same reaction with MeOH/HCl? In that case phenol, or possibly even salicylic acid might be a more convenient starting point. Even more OTC
would be the replacement of MeOH by EtOH.
Reaction is described here (available in the reference section):
Reaction of some 1,4-benzoquinone mono-oximes with methanolic hydrogen chloride (Melvyn V. Sargent)J. Chem. Soc., Perkin Trans. 1, 1982,
1095-1098DOI: 10.1039/P19820001095
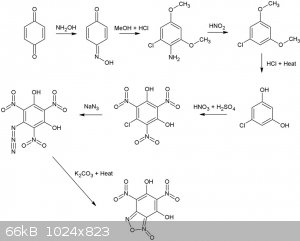
[Edited on 25-11-2015 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
@Nitro-genes,
Very nice finding that easy conversion of p-nitrosophenol (1,4-benzoquinone monoxime) into a resorcinol derivative.
In your synthesis scheme/diagram:
-you forgot the hypophosphorous acid under HNO2 (3rd Arrow); H3PO3 (taking one O away as H3PO4) but it could be substitued by ethanol (then leaving
oxydized as ethanal but this process yields some dimeric compound (biphenyls)).
-The first starting compound could be phenol and NaNO2/HCl instead of less OTC-ish p-benzoquinone and hydroxylamine  . .
-The NaN3 addition must be stoechiometric otherwise the very acidic Azido-TriNitro-Resorcinol (ATNR) will set the extra azide as HN3 free (very toxic
and gaseous like HCN).
-The last step with K2CO3 is also risky, I wouldn't heat ATNR K salt; it would be wiser to heat the water solution of ATNR free acid, thus without
K2CO3 and only add K2CO3 in the cold when the de-dinitrogenation has ended.
-Last but not least you are a NITRO-GENIUS the furoxan
ring can be formed by the azido group with the NO2 group on the right of it; thus forming the dihydroxy-dinitrobenzofuroxan you depicted; but also it
could react with the NO2 group left of it forming just the very same compound by virtue of a beautifull molecular symetry
[Edited on 26-11-2015 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks, the mechanism seems similar to a Bamberger rearrangement, though with extra substitution to a chloro rescorcinol. Very interesting reaction,
apparently, some redox involved here, amazing that the quinone monooxime (p-nitrosophenol) seems to have a higher redox potential than nascent
chlorine. Any thoughts on what the outcome might be when H2SO4/MeOH or H2SO4/water is used?
Not sure if phenol and nitrite may be a suitable starting point. Starting from phenol and nitrite results in mainly o-nitrosophenol IIRC, not sure how
this would behave during the rearrangement, compared to p-nitrosophenol. On the other hand, there is this german patent about selective formation of
p-nitrososalicylic acid from nitrous acid and salicylic. This seems controversial though, as other papers and patents suggest decarboxylation and
formation of o-nitrosophenol is favored. A closer look at the stoichiometry and conditions involved may be worthwhile.
In the paper attached they indeed use the hypophosphorous acid method for deamination (92% yield), I was contemplating whether the rearrangement and
deamination via the diazotization could be achieved in one step considering the anhydrous MeOH used. Didn't know the -ol mediated deamination resulted
in biphenyl formation and ethanal (which may also introduce by products).
Alternative to hypophosphorous may be using hydrogenperoxide (percarbonate):
Deamination of aromatic amines
US 4577046 A
The sensitivity of the compound (if it exists) is an unknown, its salts, including the potassium, may be interesting and perhaps could be used as
reasonably stable primary explosives with a likely very high density. If the above synthesis from phenol or salicylic acid could be used instead, it
could also represent an affordable and up scalable synthesis.
An alternate synthesis may be styphnic acid --> TMHI/hydoxylamine and lithium, or magnesiumethoxide in MeOH or other aporotic polar solvent (VNS)
--> 1,3 dihydroxy-2,4,6-trinitro 5-aminobenzene --> diazotization and azide substitution
Or:
3,5-dinitroaniline --> pentanitroaniline --> 1,3 dihydroxy-2,4,6-trinitro 5-aminobenzene --> diazotization and azide substitution
[Edited on 27-11-2015 by nitro-genes]
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@nitro-genes. I wondered what you wanted this paper for! We interesting idea indeed.
Let us know how you get on
I would imagine that by analogy with hydroxynitrobenzene they are are more sensetive than their hydroxy free counterparts.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Hey Boffis Saw you posted the reference paper in the reference section, thanks!
Looking at the possibility for an alternate starting point for the p-benzoquinone first. If p-nitrosphenol can be produced selectively, maybe it is
possible to cut some corners and to go directly in one reaction to 3-chlororescorcinol, although that may be pushing it.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Interesting document concerning the bamberger mechanism under different conditions, works much different than I thought. It also lists the same reaction type with MeOH and sulfuric, producing the anisole.
Attachment: Bamberger mechanism.pdf (1.2MB)
This file has been downloaded 489 times
Some further information on the decarboxylation of salicylic acid was found, although relatively little info could be found on this type of reaction.

Elimination/nitroso addition of salicylic acid using nitrous acid may (or may not) be a more conveniant route to o-benzoquinone monooximes than direct
nitrosation of phenol, though I couldn't find a a good synthesis protocol for the conversion. Decarboxylation/nitrosation of aspirin (acetyl salicylic
acid) may work even better producing less byproducts, presumably rearranging only after hydrolysis of the acetyl group. All info combined it seems
possible that nitrite mediated decarboxylation of (acetyl) salicylic acid and MeOH/Sulfuric acid mediated rearangement of the resulting nitroso
compounds may produce m-dimethoxy aniline derivatives.
Although supposedly 3,5 dinitrosalicylic acid does not decarboxylate upon nitrous acid treatment under normal conditions, it may be interesting what
the result would be after heating with 1 mole equivalent of nitrosyl sulfuric acid and trying the rearangement. The latter probably won't work though...
[Edited on 30-11-2015 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Nitrosation of phenols with HNO2 goes usually in para of the phenolic group; except if the para position is occupied; then it goes in ortho of the OH
group.
The specific case of salicylic acid probably comes from the sequence -C(OH)=C-C(=O)-
if you look closely to it:
-C(OH)=C-C(=O)- <==> -C(=O)-CH2-C(=O)-
so the transcient methylene bridge between two keto groups may nitrosate easily (this is a known fact... 2,4-pentadione turns easily into
2,3,4-pentan-trione monoxime)
-C(=O)-CH2-C(=O)- + HO-N=O --> -C(=O)-C(=N-OH)-C(=O)- + H2O
-C(=O)-C(=N-OH)-C(=O)- <==> -C(=O)-CH(-N=O)-C(=O)-
The presence of the nitroso or oxime group in alfa will favourize the decarboxylation of the beta ketonic acid (such acids are already thermally
unstable)
CH3-CO-CH2-CO2H -heat-> CH3-CO-CH3 + CO2
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Interesting indeed, wondered why salicylic acid decarboxylated so much more easily then the meta isomer, never thought this would still apply for
aromatic molecules.
Did a quick experiment in trying the decarboxylation of salicylic acid using nitrite today. Reaction was done as outlined in:
Notes - Nitrosodecarboxylation, Ronald Henry J. Org. Chem. 1958, 23 (4), pp 648–650
DOI: 10.1021/jo01098a634
The salicylic acid was dissolved in ethanol/water, after which 1 molar equivalent of sodium nitrite, dissolved in the least amount of water, was added
at once. There was immediate and strong fizzing of the solution, which only lasted a minute or so, then only weak gas production was noticed. Besides
the ethanol smell there was also a faint sweet smell, probably ethylnitrite. When left stirring at ambient temperature for a couple of hours the
solution went from colourless to a pronounced canary yellow. Upon acidifying using 37% sulfuric acid and cooling, a pale yellow precipiate resulted.
A common problem using nitrosation of phenol to produce the monooxime is that the nitrosophenol formed can react further with the phenol still present
in the solution/suspension to form all kinds of by products. Ideally, these side reactions would be absent due to the presence of the deactivating
carboxyl group. Could it really be this simple, of so why doesn't this seem to be used commercially?
In the aricle there are some holes IMO though, for instance, they perform the reaction with equimolar amounts of salicylic acid and nitrite, then
continue to make no effort of isolating the nitroso product, other than to mention a very minor amount (2% or so) of the nitrosalicylic acid could be
steam distilled (which presumably could only have come from oxidation by an excess of nitrite). Also, there is no mention of possible side reactions,
such as oxidation of the ethanol by the nitrous acid, which can result in fulminate like oxidation compounds. Starting to wonder if the CO2 is comming
from the salicylic acid or ethanol oxidation.
Maybe rerunning the reaction with only the least amount of ethanol or only using water and very long reaction times would be better? The melting
points of salicylic acid and the monooxime are too close for me to distingoush between the two, maybe someone could redo the reaction and take an
accurate melting point some day.
Another thing is that another article mentions the likely product to be a nitrosodimer. Would this still rearrange using MeOH/HCl? Are the
dimer/monomers interchangeable?
EDIT:
Dried a small amount of the resulting compound, I'm starting to wonder whether it really is p-nitrosophenol. It dissolved in water with a brilliant
yellow colour, but there is no reaction to a strong NaOH solution, nor does it explode when heated, like many MSDS seem to suggest. It seems to simply
burn, maybe somewhat more energetic than salicylic acid alone. Maybe the pure compound can be dissolved in water and measure acidity, the pKa of the
nitrosophenol should be much higher than that of any salicylic/nitro(so)salicylic acids present.
[Edited on 6-12-2015 by nitro-genes]
|
|
|
| Pages:
1
2 |
|









{kind=link}