| Pages:
1
2 |
nitro-genes
International Hazard

Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
dinitrobenzofuroxan derivatives
Hi everyone,
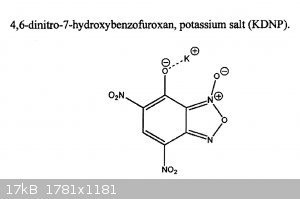
Noticed there is no thread about dintrobenzofuroxan derivatives, some of which, like KNDBF (attachment) are implemented as environmentally green
primer compositions.
I'm no chemist, but would it be possible to do a synthesis from paracetamol? Treat acetaminophen with HNO3 to introduce a nitro group in ortho
position (relative to the acetanillide group), then hydrolyzation to yield 2-nitroanilline (http://en.wikipedia.org/wiki/2-Nitroaniline). The furoxan group supposedly can be formed from oxidizing the 2-nitroanilline using hypochlorite
solution. Then HNO3+H2SO4 to nitrate this further?
Not sure where the nitro group will end up from the first nitration from acetaminophen, due to the additional hydroxyl group, which is not present in
acetanillide and in para position. ALso not sure if the second nitration will succeed.
http://www.wydawnictwa.ipo.waw.pl/cejem/vol-9-4-2012/Sarlaus...
[Edited on 24-4-2015 by nitro-genes]
|
|
|
APO
National Hazard
Posts: 627
Registered: 28-12-2012
Location: China Lake
Member Is Offline
Mood: Refluxing
|
|
From everything I've seen so far, it does look like acetaminophen usually does indeed favor addition of a single nitro group to the ortho-position
relative to the "acetanilide" group, namely if you just do a normal mixed acid nitration it will work fine[1], giving 4-acetamino-3-nitrophenol, if
you use nitrous acid, then the pH will affect where the nitro group ends up[2&3]. Hydrolysis of 4-acetamino-3-nitrophenol would then result in
4-amino-3-nitrophenol.
[1]: Possible route to resorcinol from Tylenol / Paracetamol?
[2]: Colorimetryof Serum Acetaminophen(Paracetamol)in Uremia
[3]: Nitration processes of acetaminophen in nitrifying activated sludges
Now, just to clarify, you're wondering if you could use 4-amino-3-nitrophenol in the same way you would use 2-nitroaniline to form some sort of
furoxan, which you could then nitrate, correct?
I am a bit confused because the PDF you linked has no reference to 2-nitroaniline, it only references 3-nitroaniline. Do you have a reference for
2-nitroaniline being used to form a furoxan?
"Damn it George! I told you not to drop me!"
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The synthesis of benzofuroxan from 2-nitro anilline is listed in US 4185018, they mention introducing hypochlorite solution to ethanolic solution of
2-nitro anilline. They also refer to Org. Synth., Vol. IV, page 74 ff, (1963) for the synthesis. This seems really appealing, since no obscure
chemicals seem to be needed to produce the benzofuroxan moiety itself. I haven't read every detail yet though, it was more of a late night idea that
I just had to post. :-)
Further reading: https://books.google.nl/books?id=wfJHAAAAQBAJ&pg=PA174&a...
Page 167
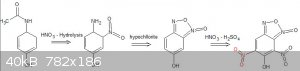
If the nitration to the dinitro compund doest work, maybe Boulton-Katritzky rearrangement using K2CO3 is possible before nitration step? Will the
furoxan group survive mixed acid treatment?
[Edited on 24-4-2015 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
The hydroxy group is the stronger inducer of orientation (vs aminoacetyl) and thus, the orientation of the nitration will be in ortho of it and not in
meta!
The resulting product of nitration with HNO3 of para-acetamino-phenol (paracetamol) will thus be the 2-nitro and not the 3-nitro.
It seems that HNO2/NO2(-) may favor the introduction of a nitro in position 3 ... this is complex chemistry and goes probably via help of the vicinal
acetamino group... nitrosation and oxydation to nitro group...or obscure nitrite addition.
To get to 4,6-dinitro-7-hydroxybenzofuroxan, one would have to start from meta-:
-nitrophenol
-aminophenol
-nitroaniline
-chlorophenol
Ortho azido-nitro-aromatics also turn upon mild heating into benzofuroxan by mutual oxydoredox of the azido and nitro groups into nitroso groups.
The synthesis scheme proposed by nitro-genes looks interesting but the para-amino-phenol (even if nitrated as 3-nitro-4-amino-phenol) is very prone to
oxydation into para-quinon-ic compounds.
I would suggest to start from 3-chloro-2,4-dinitro-phenol; then azidation with NaN3 to get 3-azido-2,4-dinitrophenol; heating to get
5-nitro-6-hydroxy-benzofuroxan and final nitration to get the desired 5,7-dinitro-6-hydroxy-benzofuroxan.
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
APO
National Hazard
Posts: 627
Registered: 28-12-2012
Location: China Lake
Member Is Offline
Mood: Refluxing
|
|
In the first link I referenced [1], it links to a patent that specifically names the compound received by mixed acid nitration using HNO3/H2SO4 as
"3-nitro-4-substituted aminophenol", which I called 4-acetamino-3-nitrophenol. Meaning that for that particular process, among others, the nitro group
is usually added adjacent to the acetamino group. Most of what I have seen shows the compound resulting from nitration or nitrite addition as having
the nitro group in the meta (3) position, however I have also seen some references as having it in the ortho (2) position. All I can really gather
from this mix of data is that you can pretty much pick whatever isomer you want, however, what you get is very dependent on reagents used and reaction
conditions.
It looks like during nitrite addition, at a pH lower then 5.5, which is apparently a deciding factor on what happens: nitrous acid generated in situ
oxidizes the acetaminophen to it's corresponding quinone-acetyl-imine, which then becomes the Michael donor, the nitrite acts as the Michael acceptor,
and Michael addition takes place. This also typically gives a higher yield, but, if OTC is the goal, just a guess since we're starting with
acetaminophen, nitrites might not be available. That's why I kind of focused on mixed acid nitration.
Both the OP's proposed synthesis and PHILOU Zrealone's sound like interesting approaches.
Are we looking at something preferably OTC and or starting from acetaminophen, or just whatever works?
"Damn it George! I told you not to drop me!"
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Many thanks for the replies, Should have been more clear in the first post: nitrites, azides, hydrazine sulfate, TCCA are at my disposal, but the idea
was indeed to do the synthesis completely OTC. I don't care for the bang of the final product, and I'm not necessarily looking for KDNP, just the idea
of synthesizing benzofuroxan derivatives from incredibly OTC chemicals seemed pretty sexy. 
So, just to sun it up, mixed acids will likely produce 4-acetamino-3-nitrophenol, but hypochlorite oxidation will likely produce benzoquinones despite
partial deactivation by the nitro group? Like said before, I'm no chemist, but I was sort of hoping the formation of the benzofuroxan group itself
might be favorable instead of oxidation. With my limited knowleadge, the furoxan group seems to "add" a lot ot stability to the ring, but could be
wrong here. How does the elimination of the NH2 group proceed during oxidation, what intermediates, similar to those leading to benzofuroxan
formation? If the hydroxy group is oxidized first, the benzofuroxan is unlikely to form probably? Another problem might be additional chlorination of
the ring by hypochlorite, but I read something about sulfite (wine making) addition to prevent this. Didn't have much time past weekend, but I will
look into it. Still hoping this may actually be a viable synthesis somehow. 
Another interesting approach is introducing a 3-amino group to 2,4,6 trinitrophenol --> US 8404897 B2. Lithium can be obtained from batteries, but
hydroxyl amine isn't really OTC (nitromethane is not easily obtained around here).
[Edited on 27-4-2015 by nitro-genes]
|
|
|
APO
National Hazard
Posts: 627
Registered: 28-12-2012
Location: China Lake
Member Is Offline
Mood: Refluxing
|
|
You're very welcome, thank you for the great idea and references. I'm far from a chemist too, but: It seems like production of
4-acetamino-3-nitrophenol, whether it be through mixed acid nitration or nitrite addition, is possible, but like I wrote before, you would need to
follow a very good procedure, very stringently, in order to receive the desired isomer.
Here is the OrgSyn prep of benzofurazan oxide from 2-nitroaniline referenced by patent US 4185018. The yields are actually very good! One should
note that the hypochlorite is added to the 2-nitroaniline in a solution of potassium hydroxide dissolved in ethanol at a low temperature. Given what
all that entails, I think that may eliminate a few of the foreseen possible issues such as chlorination of the ring or the hydroxide group inferring,
since the yield is not really being cut down by any haloform that would usually occur with ethanol, it makes me think that furoxan formation is more
favorable, and pretty quick. If oxidation still presented an issue, maybe gelatin could be used in the same way it's used in hydrazine prep?
Another approach would be to take the product after hydrolysis, convert the amine to an azide, and then heat to form the furoxan. Converting the amine
to an azide could be done in a few different ways in this case, but that lengthens the process by two to three steps, so I think we should focus on
the hypochlorite approach first.
Regarding the hydroxylamine, maybe this would work?
"Damn it George! I told you not to drop me!"
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks for looking up that reference from org syn! :-) Read the rescorcinol from paracetamol thread you linked to, seems a happy coincidence that KOH
can also be used for the hydrolysis of the amide. What temperature should the hydrolysis take place? Will the nitro group survive? The hypochlorite
oxidation using KOH/etOH may be a perfect one step procedure to invoke both hydrolysis and oxidation to furoxan at the same time.What did worry me is
that in the org syn procedure of hypochlorite oxidation, tarry compounds are mentioned forming above 10C, this may not bode well with the hydroxy
group in para position maybe. :-)
EDIT: Have extracted some 50 grams of pure acetominophen from paracetamol using boiling ethanol. Will try both the mixed acid synthesis at very low
temperatures as well as the HNO2 synthesis. I foresee big red clouds for the first approach, so small quantaties at first. :-)
[Edited on 28-4-2015 by nitro-genes]
|
|
|
APO
National Hazard
Posts: 627
Registered: 28-12-2012
Location: China Lake
Member Is Offline
Mood: Refluxing
|
|
You're most welcome! Hydrolysis can be done with either an acid or base, HCl, NaOH, or KOH will all work. References on hydrolysis of acetaminophen
and related compounds that I've seen generally cite the process as taking place in a boiling water bath or sometimes at reflux. From what I've seen,
the nitro group should not be affected. I think that the hydrolysis and furoxan formation need to be separate steps due to the temperature difference
that they're usually carried out in, and the acetic acid formed in the reaction would cause all kinds of problems. It looks like always being at or
close to 0 ℃ is very critical to prevent side reactions during furoxan formation.
Now regarding mixed acid nitration, here is the procedure for the synthesis of 4-acetamino-3-nitrophenol from chinese patent CN1966495A, re-written to be more clear, since it was not perfectly translated:
First, 80ml of 98% sulfuric acid was added to a 150ml four-necked flask equipped with a stirrer and a thermometer. It was then stirred and cooled to
below 10 ℃, at which point, 9.4g of acetaminophen was added. It was further cooled to below 10 ℃ again. Then a solution of 3.7ml of 98% sulfuric
acid and 3.8ml of 96% nitric acid was added drop wise. The addition was complete after 2 hours. Then under strong stirring, the solution was poured
into a large mix of crushed ice slurry. The precipitated yellow crystals were then filtered off and dried to give 9.2g of 4-acetamino-3-nitrophenol,
in yield of 75.6%, with a melting point of 162 ℃.
I would also advise to prepare the sulfuric and mixed acid solution before hand, and then pre-chill them in a freezer several hours prior to use. Then
to run the whole reaction cooled by a salt-ice bath, keeping the mixed acid solution addition slow enough to stay well below 10 ℃. Maybe have an
extra bath in the freezer ready encase the first one starts to warm up. You may also want to wash the filtered product with cold saturated sodium
bicarbonate solution and then with more water just to completely neutralize it. Of course a four-necked flask isn't totally necessary, I'm sure you
can use whatever setup you have as long as the reaction conditions remain the same. I'd personally like to see the nitration first, rather than
nitrite addition, since it's slightly more controversial and we've gathered more info on it, anyway, best of luck. Also post pictures!
I will also gather more info on nitrite addition, and post back here with a possible procedure.
"Damn it George! I told you not to drop me!"
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Ok, nitration went much smoother than anticipated. I did't use HNO3, I was out, and didn't want to distill something if nitrate salts could be used
instead. :-)
7.7 grams of absolutely anhydrous ammonium nitrate was dissolved in 20 ml 98% sulfuric acid at room temperature, this was put at -20., resulting a a
clear syrupy liquid. In a separate beaker, 60 ml of 98% sulfuric acid was poured and also put at -20. The next day, 9.4 grams of acetaminophen was
dissolved in the 60 ml of sulfuric acid (while on salted ice bath), only very slight exotherm occured, so addition of the AN/SA was directly done
after. Using a pipet the AN/SA was slowly allowed to drip in the SA/Acetaminophen solution, temperature did not went over 0 degrees at all times,
addition was done over the course of about 15 minutes, almost no smell of NOx was noticed, but very faint smell of acetic acid was. An interesting
note is that the solution went instantly from sort of orange to almost red with the addition of the last drops of AN/SA, which seems a good indication
for completion of the reaction. The mixture was poured in 300 ml of icecold water upon which a orange-yellow solid precipitated. This was washed with
another 300 ml of icecold water and collected to dry. It has low water solubility, stains like picric acid and sodium bicarbonate produced a bright
red solution. Looking at the solubility of the potassium salt right now, as it is in the fridge.
Some additional notes, exotherm is really manageable, salted icebath doesn't seem obligatory, crushed ice should work fine with maybe slightly longer
addition time. For the next synth I will try to minimize the amount of 98% SA needed.
The nitration also produced a small amount of sticky, chewing gum like stuff, probably left overs from the binder/sugars in the acetaminophen tablets.
Either that or polyanilline like derivatives may have formed during the reaction. It's probably better to either start with the pure stuff or do
proper recrystallization from ethanol first. (I simply evaporated everything at low temp).
Question remains which isomer we have produced, though the nitration itself seems to work. Also wonder how sensitive this stuff is to
hydrolysis/oxidation by air, with some of the entrapped acid remaining from the synthesis. Bicarbonate neutralization to produce the sodium salt may
be a better option, but how best to proceed for the hydrolysis/hypochlorite oxidation?
[Edited on 30-4-2015 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The chewing gum residues are definitely from the binder within the tablet. Any ideas for purifying acetaminophen from tablets? It is not sold in pure
form, crystallization from ethanol may be diffucult, as solubility doesn't seem to drop hard with temperature. Maybe water-ethanol mixtures couls be
used. The tablets likely contain starches, stearic acid, waxes and titanium/silicon oxides, The first two are probably hard to get rid of. Maybe using
just enough ethanol at lower temperatures can extract mostly acetaminophen. Will try different methods.
Attachment: Paracetamol solubility.pdf (91kB)
This file has been downloaded 11571 times
[Edited on 30-4-2015 by nitro-genes]
|
|
|
APO
National Hazard
Posts: 627
Registered: 28-12-2012
Location: China Lake
Member Is Offline
Mood: Refluxing
|
|
Very nice work! I was curious how it would work using a nitrate salt to substitute pure nitric acid; you got two birds with one stone. No pictures?
I'm aware I offered fairly conservative advise, you can never be too safe.
I was going to ask about the acetaminophen that you extracted, and whether you recrystallized it or not, but purity did not really sound like it would
effect the reaction very adversely. Most acetaminophen tablets also use some form of cellulose as a binder, so my guess would be that the gummy stuff
is probably mostly nitrocellulose and nitrostarch. The acetaminophen can also be oxidized to it's quinone, in which case it will either polymerize,
which could also be part of the gummy stuff, or it will still be nitrated, and go on to form the other isomer we talked about. Igniting some of the
gummy stuff would probably reveal more about what it is.
If you want to purify the acetaminophen, try to dissolve as much as you can in a solvent, filter to get rid of binders, then like you said, try to
re-crystallize. If you have a soxhlet extractor, then maybe extraction using a solvent in which those impurities have little to no solubility could
yield a more pure product. It does not sound totally necessary to me though.
Based on how you describe the nitration, I would think that the nitrated product primarily consists of the isomer we want.
The sodium bicarbonate solution was intended just to neutralize any remaining acid, not to form a salt, I totally forgot about that pesky hydroxide
group.
Regarding hydrolysis, here is a student procedure that includes hydrolysis of p-nitroacetanilide to p-nitroaniline. The piece of interest reads:
"1. Place 2.5 g of p-nitroacetanilide and 13ml of 70% H2SO4 in a round-bottomed flask.
2. Reflux the mixture for 20min.
3. pour the hot solution into 250ml of cold water taken in a beaker
4. Neutralize with 10% NaOH.
5. Cool and filter the yellow crystalline product on a Buchnerfunnel. Wash it thoroughly with water."
From what I've read H2SO4 actually works very well for the hydrolysis of acetaminophen and other acetanilide like compounds. So, you should simply
replace the 2.5g of p-nitroacetanilide with 2.72g of the nitrated product, scale procedure to work for however much product you have, then follow the
procedure up to step three. After that, you should filter to get the product, wash with cold water to neutralize it, and let it dry. Finally, weigh
the dry product.
After you do that, report back with the details of the procedure, how it went, and the results. Then I have some ideas to share on furoxan formation,
and will write a procedure to try. Sound good? Also please try to post pictures this time!
Notes:
The sulfuric acid really has to be diluted to 70%, or else sulfonation will occur in preference to hydrolysis.
I especially need the weight of the dry product (after hydrolysis) to do the stoichiometry for the furoxan formation procedure.
-------------------------------------------------------------------------------------------
Info on nitrite addition is very sparse so far, so that may have to wait.
"Damn it George! I told you not to drop me!"
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
A small amount of the ehtanol extracted acetaminophen was dissolved in the least amount of ethanol:water (2:1) at room temperature, chewing gum like
stuff remained, so the residues are from the tablets itself, it feels waxy, but does not dissolve in heptane. I think I just shouldn't have boiled the
ethanol to extract the acetaminophen, which lead to slight yellowing of the solution anyway, probably due to slight hydrolysis/oxidation of the
acetaminophen. Next time room temperature will be used.
70% sulfuric acid at reflux seems pretty harsh conditions, are you sure of this protocol? Wouldn't want something like this to happen. https://www.youtube.com/watch?v=h4pNXAtPJp8
To be honest, the idea of doing the hydrolysis in boiling KOH/etOH solution prior to hypochlorite addition seems to good to pass up. What was your
idea of the side reactions occuring with the acetate present? Formation of cyclic N,N dioxides, or oxidation of the acetate to CO2, screwing up
stoichiometry? I don't think these will occur at this temperature and pH at a rate that would give serious byproducts.
I'll devide the putative 4-acetamino-3-nitrophenol in two portions to try both methods, I want to try the oxidation to the furoxan in very small
quantities at first anyway. I have a feeling there might be tears
[Edited on 1-5-2015 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
12.5 grams of NaOH were dissolved in 50 ml water and put on ice, chlorine was produced from TCCA + HCl and bubbled through until 10 grams had been
absorbed.
1.96 grams of KOH was dissolved in 18 grams of EtOH and heated to dissolve. Then, 3 grams of the putative 4-acetamino-3-nitrophenol was added and
boiled for 30 minutes. A dark red suspension/solution developed immediately, but upon boiling, dissolution remained incomplete. At -10 degrees C,
21.75 grams of the fresh hypochlorite solution was added anyway, keeping temperature below 5 degrees. Solution did not change colour, small whiff
smelled like almonds with definite slight smell of chloropicrin. Allowing the solution to warm up after the last addition did not produce large
exotherm or lot of gas production (although bubbling could be heard), small amount acidified with HCl produced no smell of chlorine.
Seems like oxidation doesn't directly produce the furoxan, although hydrolysis may have been incomplete due to incomplete dissolution int he ethanolic
KOH, or the hydroxy benzofuroxan/potassium salt formed is soluble in the ethanol/water mixture, or the wrong isomer is produced during the nitration.
The latter seems more likely IMO, note that most of the references for production of 4-acetamino-3-nitrophenol start with O-acetylation, seems Philou
was right afterall. :-) Although I will try seperate hydrolysis step using HCl or H2SO4 as well, but i'm pretty sure now it won't work.
Too many probabilities to exclude IMO, is there a test to distinguish 4-acetamino-3-nitrophenol from 4-acetamino-2-nitrophenol?
[Edited on 1-5-2015 by nitro-genes]
|
|
|
APO
National Hazard
Posts: 627
Registered: 28-12-2012
Location: China Lake
Member Is Offline
Mood: Refluxing
|
|
Good to know you've made progress regarding the acetaminophen purification, and further perplexed me regarding what that gummy stuff is.
The use of 70% sulfuric acid may be a bit extraneous, but from what little literature I've looked at, no problems are stated before reaching 80%
concentration. It states that once you reach or go above 80% concentration, favor all the sudden massively slants towards sulfonation instead of
hydrolysis, but no immediate oxidation or charring is blatantly reported. Also keep in mind that it is not nearly as dehydrating as concentrated
sulfuric acid, and that once hydrolysis starts, the reaction begins to self dilute via release of acetic acid.
The initial assumption regarding issues that acetate could cause reaction wise was that if hydrolysis was done in situ that the formation of acetate
would drive the pH down, cause a need for an increasingly large excess of potassium hydroxide, and just seemed to add unnecessary variables to
consider. If there was not enough potassium hydroxide to compensate for the formation of the salt, as well as release of formation of acetate, then
they would form in equilibrium, or worst case, free acetic acid could react with the hypochlorite solution to form hypochlorous acid, cause oxidation,
or cause side reactions, in any case, further driving down the pH. I assumed that the pH was required to stay really high since a very concentrated
solution of hypochlorite is prepared for use, and a marginal excess of potassium hydroxide is already used. Despite all this though, the more I
consider it, the more it seems possible, as long as you have more than enough potassium hydroxide to form the salt, form the acetate, and keep the pH
very high. Excess of potassium hydroxide seems very important, if there is not more than enough for what all I mentioned, things are in equilibrium,
pH is too low, and things are not being pushed towards furoxan formation. Another modification I would make is to use just one form of hydroxide for
both halves of the procedure, that way there is not a mix of sodium and potassium ions to confuse which furoxan salt would be received.
The patent and the section of the thread I referenced do NOT start from O-acetylation, they specifically start nitration using freshly prepared
acetaminophen. However, I see your point that the ambiguity of the isomer may be causing an effect. I will look into ways to differentiate isomers,
and look into sure fire ways to get the isomer we want. Could you slow down on the experimenting a little? I think you might be moving a little too
fast and jumping to conclusions. There is a lot that goes into this to consider before going all out.
"Damn it George! I told you not to drop me!"
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ nitrogenes, I have been following your very interesting experiments with interest as I am looking at preparing 5-hydroxybenzotriazole via the
3-nitration of paracetamol, reduction to the 3,4 diaminophenol (with or without prelimenary hydrolysis) and then react this compound with sodium
nitrite to give the required triazole.
The benzene ring in the 5-hydroxy compound is more susceptable to oxidation and halogenation than the unsubstituted compound, infact according to the
literature, hypochlorite first chlorinates the benzene ring to a perchloro-4,5-diketone and then cleaves it to 4-carboxytriazole-5-(trichloromethyl)
ketone (see T Zincke JLAC 311 (1900) p276-329 [epic read for triazole fans  ]).
Given the likely similarity between the two systems is it possible that the hydroxybenzofuroxan is being chlorinated as fast as it is formed using up
the hypochlorite quickly leaving much unreacted starting material? ]).
Given the likely similarity between the two systems is it possible that the hydroxybenzofuroxan is being chlorinated as fast as it is formed using up
the hypochlorite quickly leaving much unreacted starting material?
By the way, hydrolysis with KOH in a small volume of alcohol is likely to give a ppt of the K salt of the nitrophenol that is probably going to be
sparingly soluble in alcohol. I think you would be better to use wáter and then ppt the free aminonitrophenol by carefully neutralising with HCl and
use the isolated product.
Just a thought
|
|
|
APO
National Hazard
Posts: 627
Registered: 28-12-2012
Location: China Lake
Member Is Offline
Mood: Refluxing
|
|
I was worried about the solubility of the potassium salt too, I thought about using HCl also, but not in the exact same way. Thanks for the info on
chlorination and extra insight. Nitro-genes has peaked my interest on one-pot/single step hydrolysis/furoxan formation, anyone have any ideas on how
to make it work?
"Damn it George! I told you not to drop me!"
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Haha, the mystery of the waxy substance that doesn't dissolve easily in water or heptane... I think I solved it, it's probably phlegmatized (fatty
acid bound) starch or sugar with intermediate polarity. Saw it as one of the contents in some tablet formulation. Whatever it is, it doesn't seem to
be extracted to the same extent when just enough ethanol is used at room temperature with long stirring, so that is easy.
Was worried too about the solubility of the potassium salt which seems a serious problem for the conversion to the furoxan (if at all possible). It
might be that the potassium-amino salt after hydrolysis is more soluble in ethanol than the potassium-acetamino salt, but I really doubt it. I was
maybe jumping to conclusions, but seen Boffis post and considering all posibilities, the problem is that the number of uncertainties at this point at
the various stages is too large to exclude by continuing experimentation, that is why we really need to determine somehow which isomer is formed
during the nitration.
@APO, if you recalculate the molar ratios used for the hydrolysis/hypochlorite oxidation, you'll find I already compensated the amount of KOH for the
acetate :-)
[Edited on 2-5-2015 by nitro-genes]
|
|
|
APO
National Hazard
Posts: 627
Registered: 28-12-2012
Location: China Lake
Member Is Offline
Mood: Refluxing
|
|
I see now that you compensated for the acetate portion of it, by my rough calculation, your molar ratio of KOH to putative 4-acetamino-3-nitrophenol
was ~2.28:1. Still, IF solubility wasn't an issue, and pH/concentration didn't effect the reaction too much, then molar ratio of KOH to putative
4-acetamino-3-nitrophenol would have to be at least ~3.1:1. Crunching the numbers I'm a bit confused on how you figured the amounts you used for the
last procedure.
Once I figure out how to differentiate isomers and etc, I will post here ASAP.
"Damn it George! I told you not to drop me!"
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
@ Boffis, interesting compounds these benzoriazoles, what is your plan with them? :-) How are you planning to to the reduction of the nitro-group? I
was thinking, how specific would sulfide reduction be, could this be used to distinguish between the isomers?
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ nitro-genes. My interest in the 1,2,3 triazoles is as ligands or parts there-of. I am interested in a wide variety of heterocyclic compounds and
potentially even the benzofuroxans. The hydroxybenzotriazole suffer benzene ring cleavage much more readily than the parent compound so it is possible
to make triazole-substituted glyoxalic acids in addition the the usual triazole-4,5-dicarboxylic acid I have made before.
My plan (prior to reading this thread) was to try the nitration of paracetamol with excess nitrite/acid as described in the Japan paper posted in
another thread. Hydrolyse the product with conc HCl and then reduce the resulting hydrochloride with Zn or Fe powder. I am not sure whether the
nitro-group would be reactive enough to react with say sodium sulphide but it would be worth a try; I would probably have to isolate the free base
after reduction though. One reduced to an o-diamine I would react it immediately with nitrous acid as its probably rather prone to oxidation.
Does any one know if sodium sulphide will reduce the o-nitro group in 2,4-dinitroaniline? This might offer an alternative route from chloro or bromo
benzene via the 2,4 dinitrohalobenzene and 2,4 dinitroaniline to nitrobenzotriazole. For your synth you you wouldn´t even need this reduction you
could run the hypochlorite oxidation on the nitroaniline to give 5-nitrobenzofuroxan. The nitro group will probably make the benzene ring less readily
oxidable. The nitro group could then be reduce to an amine to make the benzene ring reactive enough to nitrate further. But of course this probably
defeats the object which is to start with paracetamol!
There is a lot of interesting stuff posted on the DDNP thread and reading through some of the papers there is a fair amount of disagreement as to
which isomer is produced under different condition and so any data such as Mp is suspect. However, there is a good deal of data on on the
3-nitro-4-acetamidophenol that suggests a Mp of 218-220 C. If you plug this value into the older papers by Mendola et al you may be able to discern
the true identity of the compound they made.
Your idea that reduction of your compound may give you a clue to its identity may have some merit since o-nitrophenol are generally reducible by
sulphides while I don´t know about the m-nitro position. There is a fair bit of information on this reaction though in various threads.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Would this be useful as a very simple OTC route? Making a dough from urea and picric acid and baking in the oven?  Somewhere remote though... Anyone has full tekst? Somewhere remote though... Anyone has full tekst?
http://www.nrcresearchpress.com/doi/abs/10.1139/cjr46b-026?j...
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
There are several preparation of 2-nitroanilines via this route, 2,4-dinitroaniline from 2,4-dinitrochlorobenzene (its a patent and its on this site
somewhere, and this preparation of 2,4,6-trinitroaniline (picramide). I have been meaning to try this as I found an old German paper that desribes the
reduction of the ortho-nitro group to an o-diamine with sodium sulphide (as with picramic acid) form which I can prepare a dinitrotriazole (I hope
) but you may be able to prepare the appropriate dinitrofuroxan instead. The
CJC is usually fairly reliable.
Attached is the Can J Chem ref;
Attachment: Picramide preparation Can J Res Spencer & Wright 1946.pdf (220kB)
This file has been downloaded 1317 times
Here is the patent for dinitroaniline;
Attachment: 2,4-Dinitroaniline via urea US 1752998.pdf (70kB)
This file has been downloaded 1087 times
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks for the full text! Definitely going to try this on a small scale. Wanted to know how the reaction proceeded and was also curious whether they
would address possible sublimation issues with the picric acid at these temperatures. The synthesis of 2,4 dinitrochlorobenzene was already described
in the "pentryl" thread in prepblication, but these are a lot of steps. Synthesis of picric acid has 90%+ yield, if indeed the acetone soluble product
described in the paper is a yield of 90% TNA from TNP, this would mean an incredibly effiicent route to trinitroanilline.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The synthesis of trinitroaniline from picric acid and urea works perfectly as described. In a beaker with teflonized (gas tape) lid, 23 grams of
finely powdered recrystallized TNP was intimately mixed with 18 grams of finely powdered urea. This was (outside in the shed) heated for 24 hours at
180 deg C in a small electric oven. An extra oven temperature indicated that the temperature remained between 170 and 178 degrees C at all times. The
mix melted producing an orange liquid, that gradually became a distinct dark red. White fumes were emitted during the reaction, consisting most likely
of NH3 and CO2, probably recombining to ammonium carbonate in the air, giving the white fumes.
After 24 hours at 175 and cooling, 150 ml of water was added. It was a PITA to get it all to form a suspension, forming a glassy subtance, that needs
considerable effort to break up under water. The suspension was filtered and after drying of the resulting brown powder, was dissolved in 150 ml
acetone at 40 degrees. Brown insolubile stuff remained, while an orange solution of TNA was obtained. To this was added very slowly while stirring on
the hotplate, 150 ml water and then boiled until no smell of acetone could be noticed. This gave 17 grams TNA of a orange/yellow colour, melting point
around 180-190 ish (hotmplate, so not very acurate). :-)
I have a hunch for a OTC procedure for the selective reduction of one ortho nitro group, will post results in a few days. :-)
EDIT: This procedure is dangerous enough as it is, and only to be performed outside and with recrystallized TNP. Do not use a metal lid for the
reaction, volatilization of the picric acid was evident, creating a yellow film covering the entire reaction vessel. Contamination with metal picrates
can possibly lead to detonation of the entire mix!!! Also make sure that both the TNP and urea are finely powdered and mixed PRIOR to heating/melting.
[Edited on 11-5-2015 by nitro-genes]
|
|
|
| Pages:
1
2 |
|








