| Pages:
1
2
3
4
5 |
Upsilon
Hazard to Others

Posts: 392
Registered: 6-10-2013
Member Is Offline
Mood: No Mood
|
|
I did the number crunching first but ended up just winging it during the experiment, as the calculated volume of HCl didn't seem quite enough.
Anyway, I think it is safe to say that the substance I skimmed off of the surface is relatively pure boric acid. If I put a pinch of sodium carbonate
in the solution, it floats to the bottom and when it reaches the solid bottom layer, it goes up in a mini white mushroom cloud, my guess is that some
material is being carried up by the CO2 that is formed.
|
|
|
WGTR
National Hazard
Posts: 971
Registered: 29-9-2013
Location: Online
Member Is Offline
Mood: Outline
|
|
The solubility of Na2B4O7 is comparable to H3BO3:
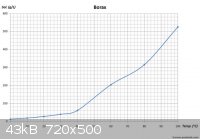
H3BO3 (g/100mL):
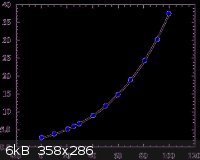
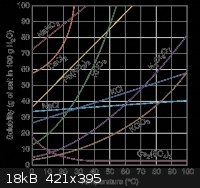
Due to the similar solubilities of these two compounds, in order to force the H3BO3 to precipitate without Na2B4O7, excess H+ should be added. This
can be accomplished by adding excess HCl. However, if too much is added, then NaCl will also precipitate, due to the excess Cl-. NaCl is much more
soluble than H3BO3 or Na2B4O7, however:
I would suggest starting with these quantities:
Na2B4O7: 0.04 moles = 8.0 g anhydrous, or 15 g decahydrate.
HCl: 1.2 mole = 120mL of 32% HCl.
Add all of this into water, forming 1 liter of solution at 20-25C.
Everything should dissolve.
Now cool the solution down to 0C on ice.
This should yield about 0.08 moles (4.9 g) of pretty pure H3BO3 precipitate, if I haven't missed something. The 0.04 moles of NaCl should stay in
solution.
After your first batch, you can successively add extra Na2B4O7 to the warm solution to retrieve more H3BO3 on cooling. Eventually NaCl will build up
in the solution, giving product that is not as pure.
I suggest making sure that everything dissolves on adding Na2B4O7 just to be sure that the system reaches equilibrium before you collect your
precipitate.
[Edited on 23-10-2013 by WGTR]
|
|
|
violet sin
International Hazard
Posts: 1482
Registered: 2-9-2012
Location: Daydreaming of uraninite...
Member Is Offline
Mood: Good
|
|
WGTR
Wiki:Na2B4O7·10H2O + 2 HCl → 4 B(OH)3 [or H3BO3] + 2 NaCl + 5 H2O
The suggested ratio was 1 x borax + 2 x HCl --> 4 x boric acid + 2 x salt( aka NaCl) so... Why are you suggesting 1.2 mol HCl for 0.04 mol borax?
That is not a slight excess. Compared to borax, NaCl solubility and boric acid precipitation would drive rxn to the right any way, no? The weak acid
sodium salt (borax)should readily hand over its sodium to the strong acid forming NaCl. unless the common ion solubility forces NaCl ppt like you
stated it stays in sol. also 0.04 compared to 1.2mol is really not advisable right? For theoretical you would only need .08 mol HCl... Your
suggested 120ml is more than twice what I recommended for one hundred grams of borax. Also there would be 0.08 mol NaCl not 0.04.
While reducing temp to ppt boric acid, NaCl isn't going to try and make a strong acid and borax again. Simple strong acid/ weak acid salt swap rxn.
I don't think even by heating to reduce volume, that you could drive HCl back out forcing borax formation again.
Your info suggests heating initial water, total dissolution of borax, addition of SLIGHT excess HCl, cooling to force B(OH)3 ppt, and filtering. Not
way over saturation of HCl.
At 0'C in 1 liter of water can retain ~25g boric acid. How ya going to get that to ppt out again? ( Sol data from wiki a few posts back boric acid
2.52g/100ml @ 0'C)
Just looked at your charts. How is >500g/l @ 100'C borax compare to 35g/l(?) @ 100'C for boric acid comparable? Side by side the curve is similar
but the scale is not even close. Really not trying to be a dick here, so don't get offended. But looks like you may have made some inaccurate
calculations or assumptions. Correct me if I'm wrong.
[Edited on 23-10-2013 by violet sin]
[Edited on 23-10-2013 by violet sin]
[Edited on 23-10-2013 by violet sin]
|
|
|
bismuthate
National Hazard
Posts: 803
Registered: 28-9-2013
Location: the island of stability
Member Is Offline
Mood: self reacting
|
|
Idea: Make boron by passing BCl3 over lithium.
How: Take an excess of Boric acid and add it to methanol then distill the trimethyl borate at 70 degrees celcius. Boil the distillate and pass the
vapor over lithium hydride. Then take the solid (LiBH4) and pass chlorine gass over it and lead the gas that will be produced over lithium with
heating. Then dissolve it in water, the insoluble material should be boron. This is harder then the idea of passing the trimethyl borate over lithium
but I'm not sure that idea will work. My references are from Wiki. Also thanks to Cheddite Cheese for help with the reaction.
[Edited on 23-10-2013 by bismuthate]
[Edited on 23-10-2013 by bismuthate]
|
|
|
WGTR
National Hazard
Posts: 971
Registered: 29-9-2013
Location: Online
Member Is Offline
Mood: Outline
|
|
Don't worry, no offense taken. It's not the first time I've made bad assumptions, or outright hair-brained conclusions. I promise I'll even make
some more.
After a bit more reading, since H3BO3 is barely disassociated in water anyway, it's likely that excess HCl won't affect its solubility very much.
That was one bad assumption that I made.
The chart for the H3BO3 is actually in g/100mL, so everything has to be multiplied by 10 to obtain g/L.
Here's what was bugging me. For the equation:
Na2B4O7 + 2HCl + 10H2O <=> 4H3BO3 + 2NaCl + 5H2O
...I saw a possible issue. At 20C, borax solubility looks like 27 g/L (0.134 mol/L). For every mole of borax, 4 moles of boric acid reside on the
other side of the equation. The solubility of boric acid at 20C is 47.2 g/L (0.763 mol/L).
(0.134 mol Na2B4O7/L) * (4 mol H3BO3/ 1 mol Na2B4O7) = (0.536 mol H3BO3/L)
Just going on solubility of the two salts alone, it looks like the equilibrium of the equation sits almost right in the middle.
Does a strong acid actually bring a fairly insoluble salt (with strong cation, weak uncommon anion) into solution, essentially pushing the reaction
towards completion? Duh...I guess it does. Carbonates, hydroxides, sulfides, phosphates, etc. In the case of carbonates, the CO2 even leaves
solution due to its limited solubility.
Ok, now I have a new project (like I don't have enough): Figure out how to calculate the equilibrium of a system like this. It bugs me that I can't
do it off the top of my head.
So...nothing to see here...just somebody smoking their socks. Move along, move along now everyone.
|
|
|
violet sin
International Hazard
Posts: 1482
Registered: 2-9-2012
Location: Daydreaming of uraninite...
Member Is Offline
Mood: Good
|
|
I enjoy reading and re-learning this stuff. Been about or greater than a decade since I was taking chem in college. The info is still there but deff
not off the top of my head. And I certainly am not the type of person who derives joy from making others feel stupid, just FYI. I WAS good at
equilibria calculations, but not any more... Sadly. Would make some of this miche easier. I plan on trying my suggestions as soon as I get back
home. Still have a lineup of other projects waiting to happen too. But it's always nice to know if ones suggestions are as cut and dry as one
assumes they are. Seems I have outlined a plan in checking the numbers for Upsilon, time for some action instead of words. I might even be able to
pull this off here( 3.5 hrs from home/lab, working) we shall see soon enough. Thanks for being a sport about the criticism
Gotta love the good 'ole mason jar experiments. Check back in later when I know how it went.
So I found some borax and muriatic acid. Basically halved the 100g borax plan. 50g borax dissolved in ~120ml boiling water, added ~26ml HCl and now
it's cooling outside. Hot water + pool acid = not fun in a small trailer. After a while ill transfer the jar to the freezer n see what comes of it.
Not going to be able to weigh it till tomorrow but visual confirmation should be easy enough tonight. Prob try a flame test to see how brightly the
sodium affects it for purity test, best I can think of right now(w/o additional chems)
[Edited on 24-10-2013 by violet sin]
|
|
|
Upsilon
Hazard to Others
Posts: 392
Registered: 6-10-2013
Member Is Offline
Mood: No Mood
|
|
I am still looking to extract boron from boric oxide without reduction with magnesium, but I need a suitable solvent to dissolve boric oxide without
reacting with it (since adding water to boric oxide reverts it back to boric acid). Would acetone or isopropyl alcohol do the trick? I want to use
aluminum foil to replace boron in single replacement, but I don't know if acetone/IPA will react with any boron formed.
|
|
|
violet sin
International Hazard
Posts: 1482
Registered: 2-9-2012
Location: Daydreaming of uraninite...
Member Is Offline
Mood: Good
|
|
I started reading this-
http://library.iyte.edu.tr/tezler/master/kimya/T000628.pdf
On page 20, near the top it starts describing some methods for elemental boron. Said the most common was magnseiothermic reduction. ~92% pure and
easy to dissolve the magnesium oxide away. Guess crystallized boron can withstand boiling HCl quite well. He(names Yelda,not sure boy or girl) then
goes on to other methods. It's a thesis paper, and he intended to reduce it with aluminum also. Worth a look.
After sitting in the cold for a while I had a nice crop of crystals in the jar. Part were initiated on the liquid surface and fell down as a whole.
So if you turn it just right the collective face reflects light from under the surface. Kinda cool looking. Would add a pic but no way to edit pics
to proper size.
Stupid auto correct
[Edited on 24-10-2013 by violet sin]
[Edited on 24-10-2013 by violet sin]
|
|
|
violet sin
International Hazard
Posts: 1482
Registered: 2-9-2012
Location: Daydreaming of uraninite...
Member Is Offline
Mood: Good
|
|
found some other papers, I know you didn't wanna do magnesium reduction, but some of these are kinda cool. each with their own difficulties. but one
uses borax directly and the other works in a similar fashion but uses the oxide, making BC4 and BN respectively. not your intended product but I found
little else on elemental boron, that wasn't covered in the link one post up. maybe fun, or useless to you
Synthesis of B4C from Na2B4O7+Mg+C by SHS Method
http://iopscience.iop.org/1757-899X/18/7/072007/pdf/1757-899...
they use high pressure argon to prevent magnesium evaporation. think it also helped efficiency. but you could probably add more magnesium.
Polycrystalline boron nitride (BN) powders were prepared by magnesiothermic reduction of boron oxide (B2O3) and ammonium chloride (NH4Cl) catalyzed
with ferric oxide (Fe2O3) at temperatures ranging from 700 to 850 ℃ under ambient pressure.
http://www.cjcu.jlu.edu.cn/EN/abstract/abstract12540.shtml
I used google translate, also i tried to download the PDF from original page, but got something about ZnO instead of the BN paper?
|
|
|
Upsilon
Hazard to Others
Posts: 392
Registered: 6-10-2013
Member Is Offline
Mood: No Mood
|
|
I may just end up doing it with aluminum reduction. Boric oxide isn't too easy to produce in quantities large enough to blindly experiment in, and I
just don't want to spend the effort creating the amounts needed to experiment with various solvents. Aluminum powder is dirt cheap and can be used in
most reduction reactions, and it won't oxidize nearly as quickly as magnesium powder. The aluminum oxide formed can also be easily dissolved by some
HCl.
|
|
|
Upsilon
Hazard to Others
Posts: 392
Registered: 6-10-2013
Member Is Offline
Mood: No Mood
|
|
From my first experiment, I ended up with a pitiful yield of boron trioxide, no more than a gram or so. I feel like the majority of this loss was in
the reaction between the HCl and the borax. I started the experiment again with 200g of borax and I added excess HCl and stirred vigorously. I am
trying to conserved as much material as possible, including that of which is dissolved in the HCl, so I am evaporating off the HCl. Just letting it
sit outside was taking way too long, plus it rained when I wasn't home so I ended up with 2x the liquid I had to begin with. I constructed a small
evaporating oven out of wood with an interior lined with aluminum foil. It uses two 60W incandescent bulbs as a heat source. It gets quite warm, to
where I can see white HCl fumes being driven off, but the evaporation of the liquid is still taking quite some time but progress is at least
noticeable.
Anyway, after it is mostly dry I am going to add HCl once again to ensure all borax has been reacted with, then I'll evaporate it off again.
Afterward, I'll do some basic solubility distillation to hopefully get a substantial amount of boric acid.
|
|
|
eidolonicaurum
Hazard to Self
Posts: 71
Registered: 2-1-2014
Location: Area 51
Member Is Offline
Mood: Hydric
|
|
This works, I have done it myself.
Required: Borax, hydrochloric acid, aluminium or magnesium powder, mixture of potassium permanganate, aluminium powder and sulphur (3:2:1), crucible,
source of heat
Dissolve the borax in the hydrochloric acid, and heat for about an hour depending on the quantity of reagent. Allow to cool, and then place in fridge
or freezer to cool further. This precipitates out boric acid, which is much less soluble at lower temperatures. Filter this off. Dry, and heat to
drive off water to form boric oxide. This will require a roaring Bunsen flame and a lot of time. Allow to cool, and grind up the resultant solid. Mix
with the relevant quantity of aluminium powder, and ignite with the mixture. Dissolve the resultant solid in hydrochloric acid, and filter off the
boron. This can be further purified and melted in an electric arc, if one can be generated.
|
|
|
Tdep
National Hazard
Posts: 519
Registered: 31-1-2013
Location: Laser broken since Feb 2020 lol
Member Is Offline
Mood: PhD is done! It isn't good but it's over lol
|
|
I actually happened to try a aluminium reduction of boron trioxide today, a ratio of 1.29:1 of oxide to aluminium powder. However, it failed to light,
despite my constant attempts with Mg ribbon and chlorates (a method that lights most thermites i've found).
Anything I'm doing wrong or is it just being stubborn? It should ignite and the reaction should be sustaining enough to continue throughout the mass?
(it was about 50g of reaction mixture)
|
|
|
Zyklon-A
International Hazard
Posts: 1547
Registered: 26-11-2013
Member Is Offline
Mood: Fluorine radical
|
|
You'll need a very fine powder, how small was the partical size?
|
|
|
Zyklon-A
International Hazard
Posts: 1547
Registered: 26-11-2013
Member Is Offline
Mood: Fluorine radical
|
|
Is it possible to isolate boron with aqueous reduction, (from Al or something), instead of a thermite? I have a lot of B(OH)3 and B2O3.
Edit: Sorry if this has been answered already, I haven't had much time to look for it...
Good yeilds are not high priority, I just want a gram or two for an element collection. I do have Al powder, but a a thermite reaction won't give high
purity products right?
[Edited on 3-1-2014 by Zyklonb]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Z:
There are no easy ways to produce boron. For an element collection, dissolve a few neodymium magnets (Nd2Fe14B) in strong hydrochloric acid. The small
amount of insoluble black/brown residue is elemental boron.
|
|
|
Zyklon-A
International Hazard
Posts: 1547
Registered: 26-11-2013
Member Is Offline
Mood: Fluorine radical
|
|
How strong does the HCl have to be? I only have about 8-9 molar HCl.
|
|
|
Tdep
National Hazard
Posts: 519
Registered: 31-1-2013
Location: Laser broken since Feb 2020 lol
Member Is Offline
Mood: PhD is done! It isn't good but it's over lol
|
|
400 Mesh aluminium powder, and the boron oxide was ground up pretty well, but I guess I can grind it more and have another go
|
|
|
Zyklon-A
International Hazard
Posts: 1547
Registered: 26-11-2013
Member Is Offline
Mood: Fluorine radical
|
|
No, that should easly be good enough, um, I have no idea why it didn't light, Mg ribbon has worked for all my thermates... I haven't tried this one
though.
|
|
|
eidolonicaurum
Hazard to Self
Posts: 71
Registered: 2-1-2014
Location: Area 51
Member Is Offline
Mood: Hydric
|
|
You are right about the difficulty in lighting, I had the same problem. Try using magnesium powder, and if that doesnt light, burning the whole
surface with a blow torch. Then, it does react, but not in a traditional thermite reaction. My pile ended up white all over.
|
|
|
Tdep
National Hazard
Posts: 519
Registered: 31-1-2013
Location: Laser broken since Feb 2020 lol
Member Is Offline
Mood: PhD is done! It isn't good but it's over lol
|
|
The ratio is 1.29:1? I'm not certain of my Stoichiometry here....
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
There's a procedure in Brauer's Preparative Inorganic Chemistry, see library.
http://library.sciencemadness.org/library/index.html
"According to Moissan, very impure amorphous boron, containing
about 80-90% B, is obtained by the reaction of B3O3 with magnesium.
According to Kroll the optimum yields are obtained as follows: A
fireclay crucible, approximately 20 cm. high and 16 cm. in diameter,
is painted with a paste of ignited MgO and sintered MgCl2 and
dried in a low-temperature oven. A mixture of 110 g. of B2O3,
115 g. of Mg shavings (the use of Mg powder frequently leads to
explosive reactions) and 94 g. of powdered S is placed in the crucible.
The reaction is started with an ignition pellet, after which it
proceeds vigorously. After the mixture has cooled, it is extracted
in water and then in dilute HCl for a week. The residue is treated
several times by heating with HF and HCl, washed with water and
dried in vacuum at 100°C. The yields are variable, with a maximum
of 46%."
Without sulphur that mixture is probably impossible to light and will not generate enough heat to self-sustain. Note that the author warns against
explosive reactions. If you want to use Mg powder try on a small scale first.
For an ignition pill, I'd recommend a small amount of stoichiom. KClO3 + Al mixure, lit with a piece of Mg ribbon. Stand well back after lighting the
Mg ribbon!
Washing the product will generate substantial amounts of H2S, so be prepared!
[Edited on 4-1-2014 by blogfast25]
Edit:
The formulation 100 g B2O3/115 g Mg/94 g S is strange though. It corresponds to 1.58 moles B2O3/4.73 moles Mg and 2.93 moles S. But acc. B2O3 + 3 Mg
=== > 2 B + 3 MgO, 1.58 moles of B2O3 require 4.74 moles Mg, leaving no Mg for the 2.93 moles of sulphur! It could explain why low yields of B are
reported.
Also, acc. NIST values of Standard Enthalpy of Formation for B2O3 ( -1273.5 kJ/mole) and MgO ( - 601.6 kJ/mole), the reduction reaction B2O3 + 3 Mg
=== > 2 B + 3 MgO would have a Standard Enthalpy of Reaction of – 531.3 kJ/mole (of B2O3), which I would have thought to be more or less enough
to make it self-sustaining, assuming you can get it to light.
If anything, I would adjust this formulation for a small scale test by reducing the amount of sulphur to 1 mole and increasing the level of Mg to 5.74
moles. The extra heat output from Mg + S == > MgS would make the total reaction heat about – 900 kJ/mole (of B2O3), similar to Classic Thermite.
[Edited on 4-1-2014 by blogfast25]
|
|
|
eidolonicaurum
Hazard to Self
Posts: 71
Registered: 2-1-2014
Location: Area 51
Member Is Offline
Mood: Hydric
|
|
Quote: Originally posted by Tdep  | I actually happened to try a aluminium reduction of boron trioxide today, a ratio of 1.29:1 of oxide to aluminium powder. However, it failed to light,
despite my constant attempts with Mg ribbon and chlorates (a method that lights most thermites i've found).
Anything I'm doing wrong or is it just being stubborn? It should ignite and the reaction should be sustaining enough to continue throughout the mass?
(it was about 50g of reaction mixture) |
Having tried to do this myself, it is almost impossible to light an aluminium/boric oxide mixture easily. I gave up on aluminium and used magnesium
instead. It worked that time. I then chucked the products into dilute acid, which dissolves away everything except the boron. I got an amorphous brown
powder.
|
|
|
eidolonicaurum
Hazard to Self
Posts: 71
Registered: 2-1-2014
Location: Area 51
Member Is Offline
Mood: Hydric
|
|
| Quote: Originally posted by eidolonicaurum | | You are right about the difficulty in lighting, I had the same problem. Try using magnesium powder, and if that doesnt light, burning the whole
surface with a blow torch. Then, it does react, but not in a traditional thermite reaction. My pile ended up white all over. |
Identical here. That, I think, is the way to do it.
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
I'm almost certain I posted this earlier in the thread, but I don't want to go searching so here it is anyway.
I prepared elemental boron starting from boric acid roach killer and magnesium powder. I heated the boric acid on a clay dish until it melted and
steam stopped coming off, to form boron trioxide. I ground this up (NOT an easy process, it's super hard) and mixed with a stoichiometric amount of
magnesium powder. I ignited the thermite, and covered the reaction after it was finished to protect it from air while it cooled. I then broke up the
'cake' of products and digested in hydrochloric acid. Everything except the elemental boron will dissolve.
It's best to avoid aluminum in this reaction because it produces some aluminum boride, which is very inert and hard to separate from your wanted
product. The magnesium analog (Mg3B2) reacts away to borane in the acid digestion step.
A link to my video on the process: http://www.youtube.com/watch?v=0QBCyOrjR2o
|
|
|
| Pages:
1
2
3
4
5 |









