Originally posted by chemoleo
Oh how annoying I can't access the patent. It requires some damn viewer, and after having spent 15 minutes on trying to get it to work, I give up.
Bloody formats. Everytime I ahve to install lots of different softwares, making the system unstable ultimately and it still doesn't work in the end!
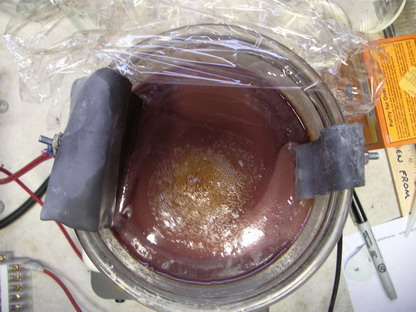
Anyway, Hilski, I imagine the formula of the manganese alum is NH4Mn(SO4)2? This is very interesting, I have never heard of this Alum to exist! I am
interested in it in particular because it may form nice crystals. The solution, after the current treatment, is it clear or is it an emulsion? It
certainly looks like it. Did you try to crystallise a small amount? What colour are the crystals? Are they stable?
|














 ....if a reaction like that might possibly have
similar usefulness
....if a reaction like that might possibly have
similar usefulness