| Pages:
1
2
3
4 |
Hilski
Hazard to Others

Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
Manganous-Ammonium alum and derivatives for use of toluene oxidation to benzaldehyde
Inspired by Cycloknight's posts in the Toluene--->Benzaldehyde thread, I decided to have a go at making the manganous-ammonium alum descibed in US
patent 808095. Basically the patent gives details on preparing manganese-ammonium sulfate from ammonium sulfate, manganese sulfate, and 60% sulfuric
acid. To oxidize the compound to the desired manganous ammonium alum (MAA), the mixture then has current passed through it in an undivided cell, using
lead electrodes. I believe, after looking at the numbers in the patent that the amount of current necessary to complete the reaction is ~0.12 amp
hours per gram of oxidant.
Using scaled down ratios from the patent, I started with the following: (Note that the amounts listed are not 100% correct, going off of the patent.
So anyone who has more patience than me can probably be much more precise in the measurements)
448g MnSO4 (2.97 mol)
204g (NH4)SO4 (1.79 mol) (The actual ratio in the patent is 2:1, but I am using about a .30 molar excess per CycloKnight)
690g H2O
1096g H2SO4 (~600ml 93% Rooto brand)
With stirring, I added the acid to about half the water, then dissolved all 204 grams of the ammonium sulfate in the water/acid mix. Then the MnSO4
was added, followed by the rest of the water. This mixture was stirred for a 24 full hours before all the MnSO4 dissolved. The yellowish precipitate
did appear. However, the overall color of the mixture was a very pale pink color instead of the orangy-yellow color I was expecting. Im guessing it
was from the MnSO4, but the drain cleaner has a bit of a brown tint to it, and that may have effected the color somewhat as well, but I'm not for
sure. The (NH4)SO4 was 99% pure and the MnSO4 was hydroponic grade, and seemed to be free from any major contaminates. It was finely powdered
similar to talc, with a whitish-pink color.
After the mixture had been stirring for over 24 hours, and everything was about as dissolved as it was going to get, I put the lead electrodes into
the jar and hooked up the power supply. I am using a computer PSU which can supply 3, 5, or 12 VDC at 30 amps. (Only 10 amps for the 12v output) I
am using the 5v output, which is actually about 4.8v across the cell. I am not using a power resistor in the circuit, since I am only using 5 volts.
I am using an old ammeter from an automobile to measure the current (all my fancy electronic measuring equipment is at work at the moment). It is
obviously not the most precise instrument in the world, but it's close enough for what I need it for. It is currently reading about 3.5 amps, and has
been holding steady there for the whole time so far. I will try to remember to bring my electrical meters home with me tomorrow, so I can get a more
accurate current measurement.
As soon as I switched on the power to the cell, a dark red color started pouring off of the anode, and within 5 minutes the entire mixture had turned
a light red color. Within 15 miutes, the mixture was a suprisingly dark red color, and after about an hour, the mixture was a deep purpleish color.
(See the photos below) Using the calculations I mentioned earlier, I figure it should take about 22.5 hours to complete the reaction. (652g oxidizer
x 0.12 amp hours per gram = 78.24 amp hours. 78.24ah / 3.5 amps = 22.35 hours) I'll take some more pictures after the 22 hours is up, so we can see
the difference in color.
Here is what the mixture looked like between 5 - 15 minutes after I switched on the power:
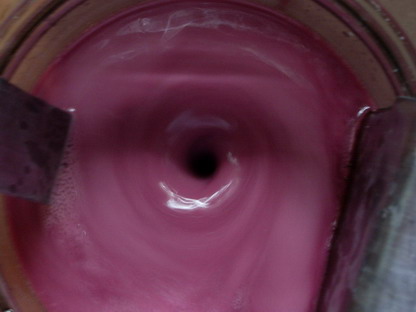

This is how it looked after an hour:


My caveman electrical test equipment; 4.88 volts across the cell

About 3.5 amps.....I think.

And last but not least........My ghetto fabulous 12v PC fan-harddrive magnet-2x4 lumber magnetic stirrer!

I'll be adding more to this in the near future.
Edit by Chemoleo: Changed title and merged threads!
[Edited on 24-1-2007 by chemoleo]
|
|
|
bio2
Hazard to Others
Posts: 447
Registered: 15-1-2005
Member Is Offline
Mood: No Mood
|
|
Nice little setup! Just a wire shunt will give an accurate current
as voltage reading on a cheapo digital meter. That way your good meters won't end up coated with sulfuric fog which inevitably seems to happen.
What are the dimensions of your electrodes and how were they made?
|
|
|
Mr. Wizard
International Hazard
Posts: 1042
Registered: 30-3-2003
Member Is Offline
Mood: No Mood
|
|
They sell a digital VOM at Harbor Freight for $3.99 that has a 10 Amp setting by changing where the leads plug in; if you have access to these cheap
tool stores.
Using a wire shunt, as bio2suggested, is a great idea, but then you have to calibrate the shunt to know what the voltage reading represents. I have a
couple of the old Wheatstone Bridges and can do that, but how would the regular amateur do it?
I'm also interested in the stirring device that looks like it was made from abiscuit fan. Any information on that would be nice.
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Nice little setup! Just a wire shunt will give an accurate current
as voltage reading on a cheapo digital meter. That way your good meters won't end up coated with sulfuric fog which inevitably seems to happen.
What are the dimensions of your electrodes and how were they made? |
Thanks. The electrodes are made out of 1/8 inch lead sheet from United Nuclear. The product name is RadMax (or something like that) and is sold as
shielding material for radioactive element storage. It's pretty cheap at around 7 or 8 bucks for a 1 square foot piece. I cut the pieces out by
cutting the surface with a utility knife, and then bending the sheet where it was cut to break it the rest of the way through. The cathode is 1.25
inches wide, and the anode, if I remember correctly, is 3.25 inches wide. The electrodes extend all the way to the bottom of the jar, which means
about 6 inches of the electrode is actually in contact with the solution.
I didn't bother checking to see what the proper current densities were for this type of reaction, beacause I figured that it didn't matter much
since this is basically a brute-force oxidation of a compound that has no possibility of being reduced at the cathode regardless. That little fact
makes this electrolytic oxidation a piece of cake compared to a lot of others, that require membranes, temperature control, voltage/current control
etc.
| Quote: | They sell a digital VOM at Harbor Freight for $3.99 that has a 10 Amp setting by changing where the leads plug in; if you have access to these cheap
tool stores.
Using a wire shunt, as bio2suggested, is a great idea, but then you have to calibrate the shunt to know what the voltage reading represents. I have a
couple of the old Wheatstone Bridges and can do that, but how would the regular amateur do it?
I'm also interested in the stirring device that looks like it was made from abiscuit fan. Any information on that would be nice.
|
I thought about throwing together a shunt type ammeter, but I didn't have the proper resistors on hand to do it. I did try a 12v 50 watt RV lightbulb
I had, and it worked. But it limited the circuit to about 2 amps, and I wanted to get at least 3-4 amps to cut down on reaction time. At 12 volts,
without the lightbulb in the circuit, the cell behaved just like Mr. Ohm predicted it would, and drew the full 10 amps of current available from the
12v output. It would have heated the cell up way too much, and would have eventually tripped the power supply (or burned it out even). So, instead I
decided to just use 5 volts, and wire in the automobile ammeter I had.
A shunt type ammeter can be calibrated by placing a trim pot in series in the shunt circuit, and placing another ammeter in the main circuit. (I know
this defeats the purpose of using a shunted voltmeter in the first place, but it's really the only feasible way for most people to do it. Maybe one
could borrow a real ammeter for a short time to use for this purpose) A load is applied to the circuit, and the trim pot is adjusted until the
shunted volt meter matches the reading on the real ammeter. The value of the trim pot would be dependant upon what range of current one would be
measuring.
Harbor Freight really is a great place to get cheap tools and equipment. I go there all the time. I work as an electrical tech for a large company,
and have more crap to measure electricity with than I can shake a stick at. I guess it's kind of ironic that I use a dirt cheap multimeter from Home
Depot when I'm at home. But I have an induction amp probe at work that would be pretty accurate for this purpose, and as soon as I measure the actual
current again, I will post it here.
As for the stirrer, it is the simplest thing you will ever build that will do the most to help you out. I used a 12v muffin fan from an old computer,
with the powerful little magnets from a computer hard drive mounted in the center of the fan. The magnets are strong enough to hold themselves to the
fan without needing to use glue, or whatever to hold them on. The plate part is made out of a piece of thick Lexan with a hole drilled in the center
of it big enough for the magnets to spin around in without hitting the sides. I only used Lexan because that's what I had on hand at the time, but
this part can be made out of anything that won't interfere with a magnetic field. A lot of folks use aluminum for this, but some have said that
aluminum bogs down the fan motor too much because of inductive loads. I can't say either way, because I haven't tried it with aluminum yet. The whole
contraption is bolted to a couple of pieces of 2x4 lumber to hold it all together. It is powered by a 120v ac/dc adapter, but anything that provides
12vdc can be used as long as it has the proper current rating. The thing works beautifully, and will stir most anything as long as proper magnetic
stirbars are used. Now I just have to figure out how to work a hot plate into this contraption, so I'll have something I can use for vacuum
disillations, or other stuff that requires heating and stirring at the same time.
Here are some links that go into detail about building homemade magnetic stirrers:
http://www.home.earthlink.net/~jsbrewing/stirrer/stirrer.htm
http://www.instructables.com/id/E570N4YH3HERXTRU8J/?ALLSTEPS
http://brewiki.org/StirPlate
|
|
|
tumadre
Hazard to Others
Posts: 172
Registered: 10-5-2005
Member Is Offline
Mood: No Mood
|
|
a good rule of thumb is that a
10 gauge wire will have a resistance of one miliohm per foot.
and make sure not to use a stranded wire because it isn't 'exactly' 10,380 cir.mills
just measure the voltage with the meter set to the 200.0 milivolt range and you get a .1 to 200 amp range.
BTW the 10 amp shunt in the meter is .1 ohm for most cheap meters, so you will lose at least 1.6 volts at 10 amps due to the resistance of the
leads+shunt.
for those chemists who commonly use molar excesses...
.0009972 ohms at 20C and will gradually reach .001114 at 50 C
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | a good rule of thumb is that a
10 gauge wire will have a resistance of one miliohm per foot. |
There haven't been too many times when I've had to make my own ammeter, but the times that I did, I always had the appropriate sized resistor. Point
being, I've never actually tried using a wire for a shunt, but I think I'll stick one in this circuit for curiosity's sake just to test the accuracy
compared to a high end digital ammeter. I'm sure it's better than my old automobile ammeter anyway. 
Also, I got to looking at the ratios in the patent again, and I think I need to add about another 110 grams H2SO4 to the mix to make it 60%. But
seeing as how it's about 4:00am, my brain cannot be fully trusted. I think I'll do it anyway. I think too much acid is better than not enough in
this case.
***Never mind. It's close enough to 60% already.
[Edited on 19-10-2006 by Hilski]
|
|
|
bio2
Hazard to Others
Posts: 447
Registered: 15-1-2005
Member Is Offline
Mood: No Mood
|
|
...... I've never actually tried using a wire for a shunt, but I think I'll stick one in this circuit for curiosity's sake just to test the accuracy
compared to a high end digital ammeter...........
Using a Fluke 87 DMM to calibrate copper wire shunts I have found that 5% accuraccy is atainable simply by measuring the corresponding wire lenght for
1, 10, or or 100 milliohms.
Then you can read the current directly throwing in the
adjustment factor if you want.
Or even better take the shunt out of one of those POS
$5.00 meters.
[Edited on 20-10-2006 by bio2]
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Oh how annoying I can't access the patent. It requires some damn viewer, and after having spent 15 minutes on trying to get it to work, I give up.
Bloody formats. Everytime I ahve to install lots of different softwares, making the system unstable ultimately and it still doesn't work in the end!
Anyway, Hilski, I imagine the formula of the manganese alum is NH4Mn(SO4)2? This is very interesting, I have never heard of this Alum to exist! I am
interested in it in particular because it may form nice crystals. The solution, after the current treatment, is it clear or is it an emulsion? It
certainly looks like it. Did you try to crystallise a small amount? What colour are the crystals? Are they stable?
[Edited on 20-10-2006 by chemoleo]
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by chemoleo
Oh how annoying I can't access the patent. It requires some damn viewer, and after having spent 15 minutes on trying to get it to work, I give up.
Bloody formats. Everytime I ahve to install lots of different softwares, making the system unstable ultimately and it still doesn't work in the end!
Anyway, Hilski, I imagine the formula of the manganese alum is NH4Mn(SO4)2? This is very interesting, I have never heard of this Alum to exist! I am
interested in it in particular because it may form nice crystals. The solution, after the current treatment, is it clear or is it an emulsion? It
certainly looks like it. Did you try to crystallise a small amount? What colour are the crystals? Are they stable?
|
You can download the .pdf format here:
http://www.freepatentsonline.com/0808095.pdf
The formula given in the patent for the alum is (Mn2(SO4)3(NH4)2SO4)
After the electrolysis, the mixture is a dark reddish brown color, and the salts are actually in a suspension of sorts. CycloKnight was able to
seperate some of the salts from the solution, and made a thick paste out of them. I'm not sure if it's possible to crystalize the alum, but the
patent states that the precursor material, manganese-ammonium sulfate can be obtained as yellow anhydrous crystals. Its formula is
MnSO.1/2(NH4)2SO4
You can probably learn much more about this compound from reading CycloKnight's posts than I can tell you. I've just began to experiment with it, and
haven't tried to use it to oxidize anything yet. I took a couple of pictures of the mixture after the electrolysis, and will post them tomorrow
hopefully.
Here's the thread with the pictures and posts made by CycloKnight:
https://sciencemadness.org/talk/viewthread.php?tid=2223&...
| Quote: | | Using a Fluke 87 DMM to calibrate copper wire shunts I have found that 5% accuraccy is atainable simply by measuring the corresponding wire lenght for
1, 10, or or 100 milliohms. |
I would think 5% is more than sufficient for this purpose, and I will definately try use this simple method in the future. I measured the current
today with a Fluke amp probe, and it read 4.1 amps, so the old automobile meter wasn't too far off the mark. I have a bunch of #10 wire, so I'll wire
in a 1 milliohm shunt after I oxidize something and need to recharge the oxidant.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Here attached is the pdf file for the patent .
This is a very interesting electrolytically regenerable oxidation reagent .
I am wondering if the oxidation product of ethanol
is acetaldehyde or acetic acid , or if either is possible
depending upon the temperature of the reaction .....
the patent doesn't say .
Attachment: US808095 Manganese Ammonium Sulfate Electrolytic Oxidation.pdf (247kB)
This file has been downloaded 2131 times
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | I am wondering if the oxidation product of ethanol
is acetaldehyde or acetic acid , or if either is possible |
Supposedly, all one needs to do is adjust the acid concentration and/or the temperature of the oxidant solution to determine whether the aldehyde or
acid is obtained as the final product. The patent does say that methanol will yield either formaldehyde or formic acid depending on the parameters I
just mentioned. I don't see why the same won't hold true for ethanol ----> acetaldehyde/acetic acid. This will definately be something I will
experiment with in the future, so if you or someone else doesn't try it first, I'll let you know how it goes.
What I like about this stuff is that it never gets used up. One could make an unlimited amount of whatever, and never have to worry about buying
anything other than reactant. (Toluene, ethanol etc)
*Edited for spelling
[Edited on 20-10-2006 by Hilski]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
This electrolytically regenerated reagent scheme should
also be applicable to other polyvalent salts which may
work as well , or better in specific reactions for particular
compounds being oxidized .
For example the ferrous / ferric sulfates , also form an ammonium alum and the precursors would be cheap as for the manganese sulfate .
More expensive would be the nickel salts .
Some of these oxidants may be regenerable intermediates in conventional electrolysis which
is done continuously in the presence of the material
being oxidized , but using the oxidant in much smaller
amount in a continuous process , and in such a scheme
the more expensive polyvalent salts would become more practical and could have certain advantages in particular reactions .
Absent the ammonium alum formation which has usefulness in this specific oxidation scheme ....there may be alternate schemes where the polyvalent
metal salt is useful alone .
And it also seems likely that the scheme could be reversed in order to arrange for reductions to be performed , probably requiring a different cell
design
or different conditions for recycling the reagent .
The metal "ous" sulfate is a reducing agent with
regards to reactivity with certain other materials ,
while the metal " ic " sulfate is an oxidizer with respect to certain other materials . So an electrolytic cell
of appropriate design and electrode materials should
be able to accomplish many different reductions as
well as oxidations , by variations of this scheme .
Another interesting point of information is that the
" ous / ic " oxidation state of the polyvalent reagent can
be transitioned purely by chemical means and no electrolysis
is absolutely necessary . For example if one has a material
which needs to be reduced , and a second different material which needs to be oxidized , and both of these materials can be efficiently reduced or
oxidized as desired by the
corresponding oxidation state of the polyvalent sulfate ....
then the reagent can be transitioned back and forth simply
by performing the reduction and oxidation reactions in the
correct sequence , using the reduction or oxidation of one
material to transition the reagent oxidation state , making it ready for use in the alternate reaction for the other material .
Ideally if one had two such precursors , one needing reduction and the other needing oxidation , the polyvalent reagent could cycled and recycled with
most or all of the
electrical requirement eliminated . Where a scenario involving a huge scaleup is involved , this would increase the efficiency and economy greatly .
[Edited on 20-10-2006 by Rosco Bodine]
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | This electrolytically regenerated reagent scheme should
also be applicable to other polyvalent salts which may
work as well , or better in specific reactions for particular
compounds being oxidized .
For example the ferrous / ferric sulfates , also form an ammonium alum and the precursors would be cheap as for the manganese sulfate .
More expensive would be the nickel salts .
Some of these oxidants may be regenerable intermediates in conventional electrolysis which
is done continuously in the presence of the material
being oxidized , but using the oxidant in much smaller
amount in a continuous process , and in such a scheme
the more expensive polyvalent salts would become more practical and could have certain advantages in particular reactions . |
There is definately a lot of room for experimentation here. Especially since there isn't much published research (that I have been able to find) on
the production and use of these types of compounds for this purpose. The good thing about it is, that the processes are fairly simple and
straightforward. And as you stated, with the exception of nickel or precious metal salts, the reagents are cheap and readily available for anyone who
chooses to experiment with them.
| Quote: | | Absent the ammonium alum formation which has usefulness in this specific oxidation scheme ....there may be alternate schemes where the polyvalent
metal salt is useful alone . |
I'm sure you are right about this. But it also has me wondering whether an alum with the manganic oxidation state can be produced by
using the same electrolytic process, only carried out in a divided cell as opposed to an undivided one? Would there even be any advantages to using a
compound such as this, over a non-alum "ic" salt?
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I take it you mean the " ous " reduced salt as opposed to the " ic " oxidized salt ? Anyway the advantage of the non-alum form is greatly increased
solubility over the alum form ,
much better contact between reactants in solution or emulsion , and a faster reaction rate ....a much more active system , which of course could be
undesirable if a gentler reaction is required , in which case the alum would be preferable .
IIRC the alum form is used for commercial reasons
because the alum is less soluble , more easily purified
and crystallized , and in storage the alum of the " ous " form is less subject to atmospheric oxidation . But the
business part of the double salt is the polyvalent metallic
salt , so the alum may be considered in most cases just
a more convenient form of the polyvalent salt having
better handling and storage properties .
It may require a divided cell or simply different cathode material or a different pH for the reduction to occur preferentially to the oxidation .
Electrochemistry is
extremely specific and even peculiar in how certain
combinations of conditions behave . It is a science
unto itself . I won't pretend to know the intricacies
of the theory on this even though I have done a few
electrolytic reactions , basically copying others work
or making slight variations .....so I very willingly defer to
others expertise or knowledge / suggestions concerning
suggested conditions and cell designs and electrode materials which would have the likely configuration needed for the reverse reaction , where the
metal " ic "
salt would be favorably * reduced * to the " ous " salt .
[Edited on 20-10-2006 by Rosco Bodine]
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | | I take it you mean the " ous " reduced salt as opposed to the " ic " oxidized salt ? |
Doh. I need to proof read myself better. Yes that is what I was intending to write, instead of what I actually did write. Thanks for the
correction.
And thanks for the lesson on the difference between alums and non alum polyvalent salts. This is all very interesting to me, and gives me something
to experiment with that could actually turn out to be useful.
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
I almost forgot. Here are a couple of pictures of the cell after about running at 4 amps for 22+ hours. The mixture has settled some, making it
appear as though there are 2 phases. It was VERY thick at that point, and the magnetic stirrer was having a very hard time. I need to get some
smaller stir bars for this, but ultimately I will rig up an overhead stirrer with either glass or nickle rods. But, for now I will be stirring by
hand for the actual oxidation reaction to make sure there is complete mixing of the oxidant with the toluene (or ethanol, methanol, glycerine, or
whatever)



|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
IIRC pure benzaldehyde is actually unstable and is subject to oxidation and polymerization unless it is inhibited with a trace of hydroquinone or
something similar as an antioxidant , and kept sealed from exposure to air . A bottle of freshly made benzaldehyde can set up solid from a
self-catalyzed polymerization probably via a peroxide of benzaldehyde which forms from exposure to the oxygen of the air acting as the catalyst .
Keeping some vapor pressure of a volatile
solvent residue or deliberately added impurity like benzene or toluene will help the storage stability
compared to the absolutely pure benzaldehyde .
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Keeping some vapor pressure of a volatile
solvent residue or deliberately added impurity like benzene or toluene will help the storage stability
compared to the absolutely pure benzaldehyde . |
I plan on doing 5 - 10 oxidations, using toluene or DMC for extraction after each. I should then have 300 - 400ml benzaldehyde in about twice as much
solvent. After that, I will wash all the extracts and the it will be stored in the solvent used to extract it until I need to use it for something,
at which time I will distill it to get the pure benzaldehyde.
Is there any reason that storing it this way would degrade the benzaldehyde? Or should I do one distillation to get the crude product and store it
that way?
I had planned on doing it this way anyway, mostly because I don't have all the glassware I need for fractional distillation at this point.
[Edited on 21-10-2006 by Hilski]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
You can store the pure benzaldehyde in a tightly capped bottle indefinitely , just add per liter , about .1-.2 gram of hydroquinone in 10-20ml of
benzene or toluene as a stabilizer and preservative if you are going to keep it for more than a week . An amber bottle and cool place are
a good idea too for long storage .
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
Just an update: This stuff does indeed work for oxidizing toluene. Sorry, but I don't have any pictures to show the reaction at the moment. The
mixture was stirred by hand, which is obviously less than ideal. But, I am currently working on an overhead stirrer which should make things a lot
more 'automated'.
I'll post more pics etc. when I have them.
*****
And one more thing I forgot. ~30 inches of #16 stranded copper wire (which I am using) gives about .01 ohms of resistance. I tapped the + wire to the
cell at two points 30 inches apart and tested the voltage drop, and the meter read .055 volts. I double checked this reading with an i1010 Fluke AC/DC
amp clamp, and it was a nearly perfect match (after moving the decimal point in the DMM reading) at about 5.58 amps. I have tested this several more
times with different voltages and currents and it is right on every time.
[Edited on 31-10-2006 by Hilski]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
An interesting potential for this cell relates to the possible
usefulness for production of pentaerythritol using denatured
ethanol and methanol as feedstocks .
If acetaldehyde and formaldehyde are formed under the same condition by oxidation of their respective alcohols
by the manganic oxidizer , then it may be trivial to make
the mixture of acetaldehyde and formaldehyde solution
needed for pentaerythritol . What I am thinking is that
a mixture of methanol and ethanol in a 4:1 molar ratio
could simply be stirred into the manganic reagent , and
then the liquid containing the acetaldehyde and formaldehyde products filtered , treated with sodium carbonate to precipitate any manganous impurity
as carbonate and filtered again . To the mixture is then
added calcium hydroxide and it is stirred and gently heated
for two to three hours , treated with dilute sulfuric acid
to precipitate calcium sulfate from the calcium formate
byproduct , the mixture filtered again and evaporated
to obtain the pentaerythritol .
If this should work as planned , it is the sort of reaction
which could be performed using five gallon plastic buckets
as reaction vessels , and would be a technically simple and cheap source for pentaerythritol .
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
Just a few pictures
Here is how the mixture looks after the reaction is complete:


If you look closely at picture #2, you can see that there is a pretty nasty emulsion. I'm not sure what this is from, but I'm hoping I will be able
to get rid of it by filtering the top layer.
Cycloknight wasn't kidding when he said the whole house smells like benzaldehyde, because it damn sure does.
***Addon: After adding about 150ml toluene to the mix and stirring a bit with a glass rod, the emulsion cleared up for the most part and I was able
to extract the benzaldehyde without too much problem. I know there is at least a little bit of benzoic acid left, because after I removed the
benzaldehyde, I could smell it in the reaction vessel. I'm guessing that its from leftover toluene from the last reaction/extraction. I doesn't
appear to have caused any problems at all, so I'm not too worried about it. I use 4 -5 amps when regenerating the oxidant, and it still regenerates
in about the same amount of time as it did originally.
Here is the really ghetto mechanical stirrer I used to stir the mixture. It worked very well, and I will continue to use it for this purpose.

-Hilski
[Edited on 6-11-2006 by Hilski]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
That stirrer sure brings back some memories for me
because it is almost identical to the first one I ever
cobbled together . And if the reaction mixture is not
particulary corrosive , what I used for a stirring shaft
was the back half or so of an old fiberglass arrow ,
using the three plastic vanes for the agitator .
The hollow arrow shaft snugly fit the shaft on the
C-frame shaded pole motor and was secured with
a drop of glue . I didn't use the second bearing that
you have there for steadying the stirring shaft itself
away from the motor , but that is a good idea especially
for higher speeds and thicker mixtures which could
cause the stirrer shaft to flex and wobble without
the extra journal there to stabilize it .
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
Possibly another (easy) oxidizer for toluene to benzaldehyde?
Here is an interesting patent that utilizes a manganic oxidizer made from oxidizing a mixture of manganese sulfate and either potassium or sodium
sulfate electrolytically in an undivided cell in 8% H2SO4. Temps for the reaction are supposed to run no higher than 30C, and it's told to produce a
high purity product with a minimum of tars and/or carboxylic acids. The example in the patent is oxidation of methoxytoluene to anisaldehyde, but
according to the author, the oxidizer should work to produce other aromatic aldehydes and acids. I find the fact that only 8% H2SO4 is needed to be
particulary appealing, along with the fact that no heating should be needed. (Large batches in plastic buckets should be feasible)
As a comparison, manganous ammonium alum needs 60% H2SO4 and 50C to react, although it does work well, and it is prepared in exactly the same way as
the oxidizer in this patent.
http://www.freepatentsonline.com/3985809.html
Sorry if this has already been posted before.
Hilski
[Edited on 23-1-2007 by Hilski]
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Looks like another good method .
I would bet that this same manganic oxidation reagent
would also be an effective reduction reagent when it
is in the reduced "ous" manganous state .
For example it possibly would reduce a phenylnitropropylene
to a phenylisopropylamine  ....if a reaction like that might possibly have
similar usefulness ....if a reaction like that might possibly have
similar usefulness 
And it would possibly also reduce nitrobenzene to aniline ,
picric acid to picramic acid .....ect.
Of course the electrical regeneration to the reduced state
would require a porous cell around the anode and plain acid
anolyte .
Anyway , here is the patent you mention above attached
Attachment: US3985809 manganic sulfate oxidizer for production of aldehydes.pdf (341kB)
This file has been downloaded 2635 times
|
|
|
| Pages:
1
2
3
4 |









