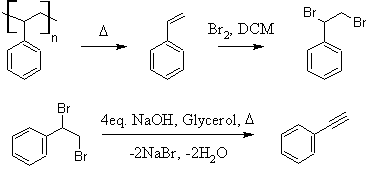Well I performed the chlorination of styrene today, and had some strange observations. The first weird observation is that it appears that styrene is
(more or less) insoluble in dichloromethane, which is clearly inconsistent with your video (a problem on my part, I'm sure). I'll describe how I
obtained the DCM and styrene, so see if you can spot any potential reasons:
The dichloromethane I obtained yesterday from paint stripper that contained a significant (~15% IIRC) amount of methanol. This I removed with two
water washes, and I'm pretty sure the methanol was all removed because I measured the density of the water from the second wash, and it was almost
exactly 1g/cm^3. I then dried it over anhydrous MgSO4, then distilled it. The first part of the distillate came over slightly cloudy, so I think there
might have been a small amount of water left in the DCM.
The styrene was obtained in exactly the manner you outlined in your video only I used plastic shot glasses with recycling number 6 and a small "PS" on
the bottom instead of cutlery. I filled a shoddy 100ml flask twice, then distilled the resulting orange liquid under aspirator vacuum, collecting
everything boiling under 100 degrees. the distillate was slightly cloudy, though.
The chlorination went smoothly, with the only unusual observation being that the dischlorostyrene appeared slightly pink. I then went ahead to strip
off the DCM under vacuum, however I went a little to far, so a fair amount of the dichloromethane was removed. I did check the mixture periodically
though, and found to my immense surprise that the DCM and dichlorostyrene had swapped! the DCM now floated on top of the dichlorostyrene, as oppose to
the other way round.
The Dichloromethane is currently sitting in the freezer, but it refuses to solidify, instead oozing about like honey.
Any ideas what could have gone wrong? I might have another crack at it later on, on a larger scale...
|