This morning, I tried something interesting... I put a few milliliters of the FeCl<sub>2</sub> solution in a test tube and a little bit of
orange iron oxide in another test tube.
Then I pipetted a little bit of conc. Nitric acid in the two test tubes. Very cool reaction:
Test tube one, with the ferrous chloride solution became a very very dark yellowish brown, similar to Ferric Chloride but even darker.
Test tube two, with the dry oxide, bubbled vigorously and quickly became an almost clear, vaguely lilac solution. The oxide completely dissolved.
So I know that I have made a solution of Fe(NO<sub>3</sub> <sub>3</sub> in test tube 2, but what happened with test tube 1? I thought the nitric guys would have kicked the ass out of the
chlorides and given me a similar Iron (III) Nitrate solution... <sub>3</sub> in test tube 2, but what happened with test tube 1? I thought the nitric guys would have kicked the ass out of the
chlorides and given me a similar Iron (III) Nitrate solution...
[snip]
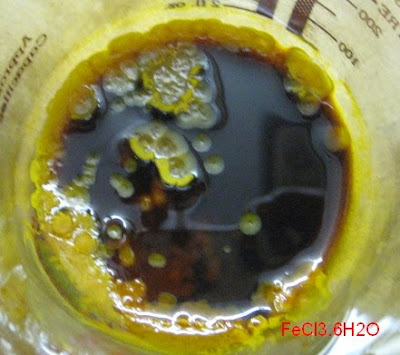
In person, the phase separation is even more distinct, and that's just overnight in a sealed jar!
Robert
|






 I just checked the contents of the test tube, and now it has become
completely transparent bright yellow!!! Would that be because of the dissipation of the NO gas?
I just checked the contents of the test tube, and now it has become
completely transparent bright yellow!!! Would that be because of the dissipation of the NO gas?

