| Pages:
1
2 |
Arthur Dent
National Hazard

Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Iron Oxide question
Hi all!
First post here, this is a great forum, chock full of great info. Among my numerous hobbies, I do a lot of electronic tinkering and some precious
metals salvaging on the side...
I've always been fascinated by chemistry, and I have been applying some of my (limited) knowledge to my hobbies.
My question about Iron Oxide is simply about advice on how to get good results making this material. I've started using the age-old recipe of letting
a few steel wool sponges sit in a mason jar filled with plain ol' bleach to obtain iron(III) oxide (Fe2O3). The steel wool was quickly cleaned off of
oils with some acetone previous to the experiment.
I will be using the Fe2O3 eventually to make some Ferric Chloride to make PCBs. I thought I might as well make my own since the stuff they sell at
electronics stores is so outrageously expensive, and concentrated HCl is so cheap at the hardware stores!
For my first attempt, I was agreably surprised by the good yield. After using a magnet to remove the leftover steel wool fillings, I was left with a
mason jar-full of brown liquid that rapidly deposited at the bottom. (see attached picture) I poured it in a large funnel with a plain coffee filter
and now its (very very) slowly filtering through.
My intention is to put the filtered material and the paper filter in my oven set at around 160 F (70 C) to dry it out thoroughly.
Or is there another method anyone recommend?
I know that there will be some carbon left along with the Iron Oxide but I believe it will be unaffectd by the HCl when I prepare the FeCl3.
If I have some leftovers, I may attempt to make a tiny bit of Thermite, since I have a large jar of fine powdered aluminum (from way back, this jar
dates well before my birth... and I' m 47!). 
Thanks in advance for your recommendations and suggestions.
Robert

|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
If you're looking to make FeCl3 from steel wool or scrap iron, dissolve it in fairly concentrated HCl (hydrochloric acid, muriatic acid), at least 20
%. Do it so that you obtain a fairly concentrated solution of green FeCl2 (ferrous chloride). This will oxidise, in the
presence of air oxygen and excess HCl to FeCl3 (ferric chloride), the active component of ferric etching solutions. It will
of course contain some excess HCl and I'm not sure whether that's desirable in a functioning etchant. If not you can try concentrating the solution by
simmering (this will also speed up oxidation from ferrous to ferric) and solid FeCl3 should at some point start dropping out at saturation. Be careful
with this step as it can hydrolyse the FeCl3 to a useless mass of oxychlorides...
With regards to drying your Fe2O3, don't do this too aggressively: calcined oxides are usually very hard to dissolve in mineral oxides like HCl...
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Thank you for your suggestion! You're right, the process you described saves me from the step of making the oxide. And I can imagine that adding some
of the iron oxide I already made to the mixture would get rid of the excess HCl and the remaining oxide would drop undissolved to the bottom. I'll
leave the resulting FeCl3 in liquid form (I have a nice large brown glass bottle that used to have FeCl3 so i'll pour it in that).
I used to have some Ferric chloride in solid form, yellowish granular (an antique bottle from JT Baker) but it's so hygroscopic that it soon turned
into a liquid just from the moisture in the air.
As for my oxide, I do have a fairly good yield from those steel wool pads. I've filtered out the water and the remaining oxide on the filter dried
into a crust that was easy to separate from the paper filter. I have about half a 80 ml becher filled with the dried oxide, i'll pop it in my oven at
70 C for about an hour or two to dry it completely then i'll crush it in my crucible to turn it into dust, I won't go above that temperature because I
don't want it to turn into magnetite LOL!
Robert
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
70 C will be a slow boat to China: you can easily go up to 150 or so before any calcination will occur. Remember that the BP of water is 100 C: below
that it evaporates only quite slowly... But even if the stuff isn't 100 % dry to the bone, it doesn't matter for your purpose.
For things like Thermite your oxide really does need to be quite dry ('keep your powder dry!')
|
|
|
NeutralIon
Harmless
Posts: 28
Registered: 19-6-2005
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Arthur Dent  |
I will be using the Fe2O3 eventually to make some Ferric Chloride to make PCBs. I thought I might as well make my own since the stuff they sell at
electronics stores is so outrageously expensive, and concentrated HCl is so cheap at the hardware stores!
|
If you are planning on etching PCB's you should consider using Cupric Chloride (CuCl2) and HCl instead of FeCl3. CuCl2 etches the Cu and produces
Cuprous Chloride (CuCl) which can be oxidized back to CuCl2 by bubbling air through it in the presence of HCl:
CuCl2 + Cl -> 2CuCl [etching reaction]
2CuCl + 2HCl + ½O2 --> 2CuCl2 +H2O [regeneration reaction]
This way you can reuse the etchant over and over, never have to make more or dispose of used etchant.
Knowledge is Good
|
|
|
watson.fawkes
International Hazard
Posts: 2793
Registered: 16-8-2008
Member Is Offline
Mood: No Mood
|
|
That's
mostly right, but not entirely. You do end up expanding your copper chloride. Eventually you'll run out of storage room and have to do something with
it. But it does mean that you don't have a disposal issue each and every time you use it.
|
|
|
ldanielrosa
Hazard to Others
Posts: 124
Registered: 25-4-2007
Member Is Offline
Mood: transparent
|
|
The beauty of CuCl2 is you can slake it with baking soda and collect the CuCO3 as parent stock for other copper salts. I'm keeping some to make
copper sulfate which I'll later convert to borate in-situ for untreated lumber on concrete (can't replace, there's a building on top).
|
|
|
NeutralIon
Harmless
Posts: 28
Registered: 19-6-2005
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by ldanielrosa | | The beauty of CuCl2 is you can slake it with baking soda and collect the CuCO3 as parent stock for other copper salts. I'm keeping some to make
copper sulfate which I'll later convert to borate in-situ for untreated lumber on concrete (can't replace, there's a building on top).
|
You can buy 99% pure CuSO4.5H2O OTC as root killer [Zep is one brand.]
I use that along with CaCl2 [ice melter] to make CuCl2
Knowledge is Good
|
|
|
peach
Bon Vivant
Posts: 1428
Registered: 14-11-2008
Member Is Offline
Mood: No Mood
|
|
You can use sulphuric with peroxide for PCBs can't you?
I seem to recall hearing about a guy doing that and loving it over the solid ingredients, saying he could just top the bath up as it ran out. It's
also the first stage in making copper chloride, to produce the sulphate with sulphuric and peroxide, then treat it with hydrochloric. Since the
hydrochloric isn't oxidizing and won't attack the copper alone.
I have a tub of ?sodiumpersulphate? crystals, sold as fine etch crystals at Maplin, RS, Farnell etc. They work nicely. The solution remains a pale
blue colour and I can easily see how the board is doing. It also etches gently, so there aren't pinholes and wrecked tracks all over the place.
Ferric chloride, I can't see through and it has a tendency to pinhole / destroy tracks, it also stains things and is generally nasty. It does go gooy
/ liquid if left open yep. And is also extremely expensive from the electronics places for what it is. If it was anhydrous, the price might be a bit
more reasonable.
If you're in the UK, I can send you a scoop of the fine etch stuff to have a go with on a small PCB if you like. I have about a kilo of it and the
chloride I think. A kilo of fine etch crystals makes around 5l of solution.
{edit}I had a VERY ROUGH go at drying some PCB chloride at one point under hydrogen chloride. It's quite funny, because it melts far below what you'd
expect something like that to, and then decomposes when boiling if not under HCl(g). It, like it's other friends (BF3 / AlI3 / AlCl3 and so on) all
function as Lewis acids and tend to do something similar, subliming, hygroscopic, decomposing etc.
{double edit}Thanks for those links in the PM about turbines Watson, I'm sorry I didn't say that earlier.
[Edited on 29-10-2010 by peach]
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
@ peach: I wasn't aware of that H2SO4/H2O2 process. I'll have to try that.
The only other PCB etching process I ever used was ammonium persulfate, and I didn't like it very much because it did a pretty aggressive job on the
etch mask and I ended-up with half-eaten traces and missing spots.
I much prefer the FeCl3 process because i've been succesful 95% + at producing nice PCBs, and after washing them off, I just drop 'em in a solution
from MG chemicals called "Liquid Tin". Fantastic results and nice clean PCBs, whether they're brown bakelite or green epoxy glass. But you're right,
it's a mess to clean up and it's a pretty intense oxidizer (keep your tools away or they'll get that nice brownish look)
Robert
|
|
|
peach
Bon Vivant
Posts: 1428
Registered: 14-11-2008
Member Is Offline
Mood: No Mood
|
|
I have a bunch of old inkjets lying around.
I've seen a guy who's managed to convert one, and I will have a go when I get a chance, to print PCBs directly, without the transfer papers or
photoresists.
It needs the roller swapping to a table and the ink swapping to a permanent marker kind.
Imagine how darn cool that'd be!? To just click print in eagle or pcb express and have it zip out on the printer.
Then it'd just need a dremel fitting as one of the cartridges and oh yeah!!!! Masked and drilled all in one!
I bet loads of guys would like one of themZ.
Combine it with a toaster reflow and that's DIY prototyping alright!
[Edited on 29-10-2010 by peach]
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Ferrous Chloride
After Blogfast25's recommendation, I have tried the trick of dissolving steel wool pads directly in highly concentrated HCl. All I can say is that it
worked marvellously! Below are the steps to my FeCl2 experiment:
I started off with my proverbial mason jar filled with 8 steel wool pads, this time I used "extra fine" steel wool (image 1) so that it would
dissolve faster.
I put the jar in a cold water bath, just in case the reaction is very exothermic. All of this is done outside of course.
Then, I poured about 200 ml of concentrated (32%) HCl, just about over the top of the steel wool. Within seconds, the reaction was vigourous and the
steel wool seemed to dissolve very fast. I added a little shot or two of H2O2 which seemed to increase the reaction even further. The boiling liquid
was brownish yellow. After the reaction slowed down, I removed it from the water bath.
After leaving the jar outside overnight (0 celcius outside), there was a very sharp separation of light green crystals that settled at the bottom of
the jar (a good centimeter's worth - image 2) with the remaining acid on top. I poured out the acid which left the crystals, which I dissolved in 200
ml boiling distilled water. Then I filtered the contents of the jar. The resulting liquid is a very bright lime green, (image 3) which I figure is a
fairly pure solution of FeCl2, with a bit of excess HCl in it.
Would the addition of H2O2 oxidize the FeCl2 to FeCl3, or should I just leave time do its job and expose the solution to open air to make it oxidize?
When I disposed of the remaining acid, I added a liter of water and some sodium bicarbonate to neutralize it until it didn't fizzle anymore. There was
a fair amount of Fe in the acid left because as I was adding the bicarbonate, the solution turned murky brown.
So there I am with my bottle of FeCl2 solution, ready to turn this into FeCl3 eventually. I have to admit it is a beautiful color right now but it is
still very acidic (a piece of PH paper turns bright magenta/violet).
The solution is stored in an acid-safe HDPE bottle.
Ref: http://www.progmontreal.com/arch/fecl2.jpg
Robert

[Edited on 20-11-2010 by Arthur Dent (typos)]
[Edited on 20-11-2010 by Arthur Dent]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Good work and good reporting!
You can simply oxidise your FeCl2 by putting the solution in an oven or heating it in open air with calor gas or something similar. But even at room
temperature it will oxidise fairly quickly, provided air can get to it (stir frequently). I made some solid FeCl2.4H2O a few weeks back with the aim
of turning it into very concentrated or solid FeCl3 (.6H2O). Well, yesterday I uncovered it and a lot of it had already oxidised: covered and not even
in solution! Ferrous chloride is almost impossible to keep in the +II oxidation state…
You need however (and this is important) to keep the acid reserve of the solution high, otherwise the stuff oxidises to (hydr)oxy chloride or even to
oxide (Fe2O3.nH2O). That’s no good for etchant solution.
The overall reaction (with oxygen) is:
FeCl2 + 1/4 O2 + HCl FeCl3 + ½ H2O
You can see it needs HCl.
Here’s what I did. I dissolved my solid (but partly already oxidised) FeCl2 in a minimum of HCl (20 % in my case) and heated it up. The hot solution
can be as concentrated as 50 w% (you need 30 – 50 % for ferric etchant). Then I oxidised it using nitric acid. That step is not strictly speaking
necessary, but it’s quicker. If you’re not used to using nitric, don’t start now. Just keep the solution heated and stir
frequently, it will oxidise.
If you’re gonna use peroxide, be very careful applying it to a hot concentrated solution of FeCl2: it may react quite violently, so go very slowly.
Or add it slowly to a cold solution and then heat slowly and carefully… The risk is mainly overheating of the solution due to released reaction
(oxidation) heat…
Now you should have a fairly concentrated solution of FeCl3. If it’s not concentrated enough, gently simmer it in. I did this yesterday and it was
uneventful. I simmered to almost nothing and on cooling got some solid FeCl3.6H2O too. This product is very hard to actually crystallise because
it’s so damn soluble in hot HCl!
You should be able to have a good idea of concentration by accounting: the amount of Fe used and the total weight of the obtained solution allow you
to accurately estimate the strength of your etchant solution. It may also be wise to filter it once before use: coffee filter should probably do it
just about…
[Edited on 20-11-2010 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
The last few days crystals of FeCl3.6H2O have started to form from my (super?) saturated solution. They’re slowly growing day by day. It’s cold
here so that doesn’t favour crystal growth and I didn’t use any seed crystals to start with. You can see the small round structures that grow
radially from a nucleus…
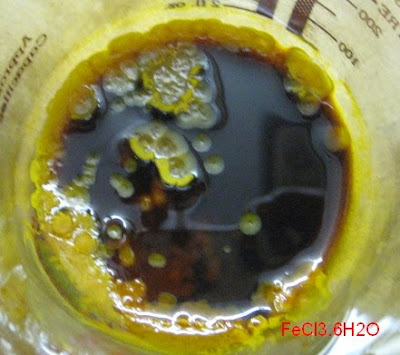
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Looks great, blogfast25!
I'll do my FeCl3 by the way of peroxide as you suggested, I'm overly cautious with HNO3, even though I occasionally make Aqua Regia with it.
Then i'll slowly concentrate it to a deep red solution with a little heat, just like the stuff you buy at the electronics store.
Congrats on your upcoming lewis acid! 
Robert
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
@Arthur: thanks, but hydrated FeCl3 isn’t a Lewis acid: only anhydrous FeCl3 is. And AFAIK, FeCl3.6H2O can’t be dehydrated with the usual
trickery…
FeCl3 in solution is a weak Bronsted acid:
[Fe(H2O)6]3+ + H2O [FeOH(H2O)5]2+ + H3O+
With acid constant K = {[[FeOH(H2O)5]2+] x [H3O+]} / [[Fe(H2O)6]3+]
… because the central electrical field of the Fe3+ ion is strong enough to expel one of the protons of its water ligands. Aqueous FeCl3 solutions
therefore always react slightly acidic due to hydrolysis.
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Reviving this old thread of mine.
I've been preparing some more Fe<sub>2</sub>O<sub>3</sub> and FeCl<sub>2</sub> recently, using the same methods
outlined as above. However, i have have slightly different result for both chemicals.
While preparing some iron oxide, I used some "Bulldog" steel wool and bleach (5% solution of sodium hypochlorite). But instead of the dark brown
precipitate that I obtained (see 1st photo of this post), what I got was a pitch black precipitate. Not a trace of brown. Since the result was
different, I figured that the steel wool was a different brand, and might have a higher content of carbon? Also, I left the mixture for a good month
outside in the shed.
Could it be possible that I made Fe<sub>3</sub>O<sub>4</sub> instead of Fe<sub>2</sub>O<sub>3</sub>?
The dried-out precipitate is black.
EDIT: I added some HCl to the black iron oxide and I got the very nice color of Ferric Chloride:

As for the Ferrous Chloride, I used the same Bulldog wool pads and 32% HCl, and it fizzed vigorously, but this time I did not add any hydrogen
peroxide to the solution, so instead of a bright lime green solution, I have a light mint green solution, so I guess that my initial attempt yielded a
mixture of Ferric and Ferrous chloride because I oxidized a bit of the FeCl<sub>2</sub> into FeCl<sub>3</sub>, hence the lime
green color. This second attempt yielded a more pure solution of FeCl<sub>2</sub>.
So with that pure solution (twice filtered), could I synthetize another interesting compound? I don't think that I can make the acetate out of the
chloride, and anyway, I have very little precious glacial acetic acid left. But I recall that the nitrate ion would displace the chloride ion, so if I
add (cautiously) some nitric acid, I should get a solution of ferric nitrate, should I?
If anyone has an interesting synthesis to suggest with FeCl<sub>2</sub>, please do!
Robert
[Edited on 9-4-2011 by Arthur Dent]
--- Art is making something out of nothing and selling it. - Frank Zappa ---
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
If it’s Fe3O4 (Magnetite) it should be paramagnetic (but you might need a neomagnet to prove that, it’s not like pure iron!) Dry some, grind it up
and test the powder on a white piece of paper and a strong magnet beneath itL it t=you can make it move it contains Magnetite...
I think you’re reading too much into the colour though: ‘lime green’ and ‘mint green’ sound like concentration differences to me, not
degrees of oxidation. Yellow Fe3+ and green Fe2+ makes for a dirty ‘khaki’ anyway. FeCl2 solutions are fairly stable, as long as you don’t go
and actively blow air through them… I’ve got some stainless steel solution (in HCl) in a closed, clear bottle and it doesn’t change colour: no
O2, you see.
An interesting compound of Fe2+, and one that is really stable in air (no oxidation, whereas solid FeCl2 hydrate does oxidise around the edges), is
Mohr’s Salt. That’s an ammonium sulphate/Fe(II) sulphate double salt, hydrated. It forms nice lime green and very well formed crystals. Normally
you’d mix a concentrated solution of both salts and then ice, and Bob’s your uncle because the salt is poorly soluble in cold water and
crystallises out. But it’d be worth trying with FeCl2 and an excess of ammonium sulphate (from the garden centre!), like:
FeCl2(aq) + 2 (NH4)2SO4(aq) + 6 H2O(l) === > (NH4)2Fe(SO4)2•6H2O(cr, cold) + 2 NH4Cl(aq)
Mohr’s Salt is nice to grow big crystals with; a bit like aluminium and iron alums.
Mohr's Salt is used in analytics to prepare stable standard solutions of Fe2+ with.
]
[Edited on 9-4-2011 by blogfast25]
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Yup confirmed. It's Magnetite, the oxide was travelling on the inside of the plastic jar as I was moving around a Neodymium magnet on its surface.
The Ferrous Chloride was created with only 32% HCl and the iron wool pads (to excess) so I guess I cannot come up with a more concentrated solution
that way. Maybe a stream of HCl gas through the steel wool would have provided an anhydrous version of the compound buti'm totally not equipped to
deal with HCl gas (and don't want to!)
The Mohr's salt sounds interesting! I just might try that. And Bob's not my uncle, he's my evil twin brother...

Robert
--- Art is making something out of nothing and selling it. - Frank Zappa ---
|
|
|
m1tanker78
National Hazard
Posts: 685
Registered: 5-1-2011
Member Is Offline
Mood: No Mood
|
|
Robert, I may have missed something but you want ferric chloride to etch PCB's? If you use the toner mask method then all you need to etch the PCB is
HCl and H2O2. I've done 20mil traces with equal spacing before without a problem.
Tom
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Thanks Tom,
Yeah I never tried that. Maybe i'll try that out next time I etch a few PCBs.
For now, i've been using pre/etched, pre-drilled standard dot pitch protoboard PCBs with point to point connections, because i'm a lazy bastard! LOL!
Returning to the ferrous chloride, no this batch won't be used for ferric chloride, i'll use it to prepare some other iron compound, just for the kick
of it...
This morning, I tried something interesting... I put a few milliliters of the FeCl<sub>2</sub> solution in a test tube and a little bit of
orange iron oxide in another test tube.
Then I pipetted a little bit of conc. Nitric acid in the two test tubes. Very cool reaction:
Test tube one, with the ferrous chloride solution became a very very dark yellowish brown, similar to Ferric Chloride but even darker.
Test tube two, with the dry oxide, bubbled vigorously and quickly became an almost clear, vaguely lilac solution. The oxide completely dissolved.
So I know that I have made a solution of Fe(NO<sub>3</sub><sub>3</sub> in test tube 2, but what happened with test tube 1? I thought the nitric guys would have kicked the ass out of the
chlorides and given me a similar Iron (III) Nitrate solution...
Ooh the magic of chemistry! I discover new stuff every day!
Finally, here's a cool picture, showing the oxidation of the top layer of FeCl<sub>2</sub>

In person, the phase separation is even more distinct, and that's just overnight in a sealed jar!
Robert
--- Art is making something out of nothing and selling it. - Frank Zappa ---
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Ok, I wrote down the formula and i'm not sure if I'm right but could it be possible that in my test tube 1 experiment, I completely oxidized the
Ferrous Chloride into Ferric Chloride?
3•FeCl<sub>2</sub> + 3•HCl + HNO3 => 3•FeCl<sub>3</sub> + 2•H<sub>2</sub>O + NO
Sorry, I'm not a wiz at figuring out what one ion does with another... but would that sound about right? But then, I'm not sure if there was much HCl
left in the jar above, since the remaining iron wool did not react at all.
EDIT:  I just checked the contents of the test tube, and now it has become
completely transparent bright yellow!!! Would that be because of the dissipation of the NO gas? I just checked the contents of the test tube, and now it has become
completely transparent bright yellow!!! Would that be because of the dissipation of the NO gas?
Robert
[Edited on 10-4-2011 by Arthur Dent]
--- Art is making something out of nothing and selling it. - Frank Zappa ---
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Arthur Dent |
This morning, I tried something interesting... I put a few milliliters of the FeCl<sub>2</sub> solution in a test tube and a little bit of
orange iron oxide in another test tube.
Then I pipetted a little bit of conc. Nitric acid in the two test tubes. Very cool reaction:
Test tube one, with the ferrous chloride solution became a very very dark yellowish brown, similar to Ferric Chloride but even darker.
Test tube two, with the dry oxide, bubbled vigorously and quickly became an almost clear, vaguely lilac solution. The oxide completely dissolved.
So I know that I have made a solution of Fe(NO<sub>3</sub><sub>3</sub> in test tube 2, but what happened with test tube 1? I thought the nitric guys would have kicked the ass out of the
chlorides and given me a similar Iron (III) Nitrate solution...
[snip]
In person, the phase separation is even more distinct, and that's just overnight in a sealed jar!
Robert
|
There’s something you need to know here: Fe2+ forms nitrosyl complexes with NO: e.g. the dark coloured Fe(NO)(2+) (one iron ion complexes one NO
molecule), similar to the better known carbonyl (CO) complexes (so important to nickel chemistry). When you oxidise Fe2+ with nitric acid, the
by-product NO can lead to strange colours, depending on exact conditions of concentration and temperature. The NO complex isn’t stable at higher
temps. and the NO gets blown off. But you’ve oxidised the Fe [+II] to Fe [+III] alright! 
| Quote: Originally posted by Arthur Dent |
3•FeCl<sub>2</sub> + 3•HCl + HNO3 => 3•FeCl<sub>3</sub> + 2•H<sub>2</sub>O + NO
Sorry, I'm not a wiz at figuring out what one ion does with another... but would that sound about right? But then, I'm not sure if there was much HCl
left in the jar above, since the remaining iron wool did not react at all.
EDIT: I just checked the contents of the test tube, and now it has become
completely transparent bright yellow!!! Would that be because of the dissipation of the NO gas?
Robert
|
Your reaction equation is correct. But redox reactions can be tricky to balance in one go. To make it easier we tend to split it up in the
half-reactions: one the oxidation:
FeCl2 + HCl === > FeCl3 + H+ + e- … (eq.1)
And one the reduction reaction:
HNO3 + 3 H+ + 3e- === > NO + 2H2O … (eq.2)
To balance, multiply (eq.1) by 3, so the number of electrons match (otherwise your overall equation cannot be electrically neutral!) and add to
(eq.2): that gives you your equation! (The 3 e- and 3 H+ on both sides drop out)
And yes, your NO has flown the nest, leaving behind the lovely yellow FeCl3…
[Edited on 10-4-2011 by blogfast25]
|
|
|
White Yeti
National Hazard
Posts: 816
Registered: 20-7-2011
Location: Asperger's spectrum
Member Is Offline
Mood: delocalized
|
|
I'm surprised that you guys try to make iron oxide on purpose. I used to make it by electrolysing salt water with iron nails, but stopped in favour of
letting scrap iron rust in salty water over several months. I got ~120g by this method without doing anything at all. I encourage other people to do
the same.
If you use galvanised iron or steel, you need to get rid of the zinc layer by reacting with vinegar or HCl, the choice is yours.
Here are some pictures if anyone is interested. It's not pretty, but it works.

My current stock of iron oxide...

One of my containers (contains about 1L of salt water and ~500g of iron metal slowly rusting away). I have four containers of all shapes and sizes:

Inevitably, the containers are permanently stained with rust, as seen here:

Good things come to those who wait
"Ja, Kalzium, das ist alles!" -Otto Loewi
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
LOL, brings back memories, this was my 1st post on SM!
Indeed, an interesting alternative, and the results provide a good quantity of Iron Oxide, but the technique of using steel wool with bleach also
works, just a little bit faster. As for your glass containers permanently stained with rust, just dip them in equal parts of HCl and water, you'll be
surprised at how fast they will be back to their original pristine, sparkling condition!
I'll try this technique of yours on a few steel wool pads, or maybe an old soft iron core transformer. Do you leave them in open air or do you enclose
them to avoid evaporation?
Robert
--- Art is making something out of nothing and selling it. - Frank Zappa ---
|
|
|
| Pages:
1
2 |









{kind=link}