RogueRose
International Hazard

Posts: 1593
Registered: 16-6-2014
Member Is Offline
|
|
Growing crystals under vacuum in a thermos (very slow temp drop) & seperating mixed salts?
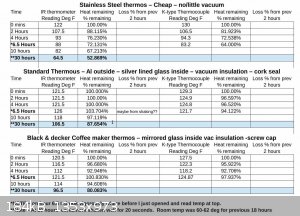
I've been testing how long some of my thermos's can retain heat and was pleasantly surprised when I found that after 30 hours, one unit had only lost
12.5% of the heat and the other about 20%. IDK if it has anything to do with having a starting temp of 130F and room temp being 70F for the first 15
hours then 60 for the second 15 hours.
In each of these I could easily use a silicone or rubber stopper and attach a vacuum pump to reduce the pressure. I would think that this would cool
the liquid faster, at least initially, but IDK how that would work or if there are any benefits to doing so as opposed to allowing the temp to just
fall slowly.
I'm going to be repeating this with boiling water to see how fast the temp will drop with the 130F I started with. I'm guessing it may drop faster at
first as the exterier of the bottle is a larger "sink" or temp difference.
I have some KCl/NaCl that is supposed to be 95/5 mix that I would like to remove as much Na from as possible. I thought this may be a good way to try
to slowly drop the temp allowing for only KCL crystals to grow (the solubility is almost 2x NaCl at boiling point). Then when the temp gets to the
point where the solubility lines cross, or slightly before, then remove the KCl crystals.
Any other salts that might be especially interesting to try in this or salts to try to seperate this way?
|
|
|
semiconductive
Hazard to Others
Posts: 325
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
| Quote: | Quote: Originally posted by RogueRose  |
In each of these I could easily use a silicone or rubber stopper and attach a vacuum pump to reduce the pressure. I would think that this would cool
the liquid faster, at least initially, but IDK how that would work or if there are any benefits to doing so as opposed to allowing the temp to just
fall slowly. |
|
For recrystallization, often impurity atoms change the aspect ratio of crystal growth; this is compounded with how quickly the crystals grow. The
slower a crystal grows (percentage wise), typically, the more pure and regularly shaped it will be. In crystal growing competitions, often the
hardest part is getting the initial seed crystal to be the only one and highly pure. Typically, this is achieved by controlling the super-saturation
of the solution and allowing it to drop very slowly in temperature at first. Additonally, the chamber is typically kept in the dark, very still (no
shaking) and at a very even temperature for days just to start the crystal growth. The process becomes less critical once the initial crystal is
formed.
There's a few issues with using a vacuum pump. The first is that water's energy of vaporization actually rises as the temperature drops. eg: The
energy lost per mole of water actually increases as the water temperature drops. Depending on whether your vacuum is constant pressure or constant
volume loss/minute, you may actually end up cooling the water faster as the temperature drops.
The second issue is that Silicone has around 1 micron holes in it. In general, it's a non-polar substance so water or polar molecules don't want to go
through it. However, plain old air will actually work it's way through silicone fairly quickly. So, even though rubber is less chemically inert ...
true rubber is actually a better seal than silicone for vacuum applications.
To keep a thermos bottle at an even temperature, while loosing water vapor; there is a relatively easy way to increase the insulation effect while
removing vapor. Putting two concentric tubes (Horizontal/spiraled) into the thermos bottle cap and insulating the tubes outsize the thermos allows
air to be sucked in the outer tube, while water vapor is pulled out the center tube. (You could even coil them inside a second thermos bottle
allowing the water condensate to collect in the second thermos. ) The result is that a significant portion of the heat of vaporization will be
transferred from the liquid in the center tube into the air in the outer tube. Think insulated allihn condenser ... as the water vapor condenses in
the center tube, the heat is transferred to the air in the outer jacket. The longer the allihn condensor, the higher the total heat transfer will be
and the lower the total heat loss. The only important issue is that air in the outer tube move in the opposite direction than water vapor in the
inner tube. You don't even need a vacuum pump ... but a simple aquarium air pump to pressurizing the outer tube will have a similar effect.
[Edited on 21-1-2018 by semiconductive]
|
|
|
RogueRose
International Hazard
Posts: 1593
Registered: 16-6-2014
Member Is Offline
|
|
Purifying 95%/5% KCl/NaCl mix via recrystallization using different saturation points
I've tried to do this without a thermos but may use that for other salts like CuSO4, FeCl2, Cu(NO3)2, etc
I've done some extractions of the KCl that look promising.
I'm a little worried that I'm not going to be able to purify a mixed K/Na chloride mix as I've heard that the two can make a "hybrid" crystal of the
two creating a cube like structure, but I think there needs to be certain amounts of each (possibly equal..?) to make this possible.
Since KCl has a much higher solubility rate at higher temps, I thought I'd use that to my advantage by making a super saturated solution in boiling
DH2O, decanting the solution (through filter removing undissolved particles), then allowing it to slowly cool (about 12-24 hours) to about 50-60F.
NaCl has a solubility of about 314g/L from O-100C while KCl is something like 280-560g/L from 0-100C
The mix is supposedly 95% KCl and the remaining is NaCl so at a saturated 1L solution should have about 560g KCl and 28g of NaCl. Since the NaCl
should not fall out of solution until the concentration reaches above the .314g/ml then there is a lot of room to keep the NaCl in solution.
I would like to re-use the brine from which the the crystallized KCl was extracted, but it will have some NaCl already in it, so I'll have to keep
track of what the possible concentration is and stop using it when it reaches a certain point.
I figured I could add ~300g of the salt mix being 285g KCl and 15g NaCl, heat and cool, then extract crystals again. This will leave behind the 15g
of NaCl for a total of about 43g of NaCl in the solution from the two processes. I suspect I can repeat this until a certain point where NaCl starts
to fall out of solution with the KCl, but I'm not sure where that point might be and how to identify that.
now the pics of the crystals I have, the ones on top are the ones that formed last (from about 90F to 50F) and they are nice beautiful needle like
crystals, which seems to identify with KCl though I thought I've also seen cubic crystals of KCl (I know NaCl can make cubic ones)
I pulled all the remaining liquid off with vacuum then heated at about 250F for a few hours and it turned into a hard rock with some odd looking
"powdery crystals" moving up the side of the container. IDK if these crystals are still pure KCl or if they may have higher concentrations of NaCl in
them (as this may be from the remaining liquid which has some NaCl). I have kept this "creeping salt" separate from the rest of the salt extracted.
I guess I should have washed the KCl with some cold water, but I plan on at least one more recrystallization which should remove any NaCl that carried
over). IDK any other way to wash the KCl crystals that doesn’t' dissolve a good bit on contact with the water/solvent. Maybe methanol would wash
off the liquid solution mix and that could be discarded or distilled to recover the methanol? would an 50/50 alcohol/water mix (-15 to 0 C in temp)
work as well or would the water in the mix still dissolve some of the KCl crystals?
Would there be any difference in the level of purity in the crystals that have formed in layers as it has slowly cooled from 212 to ~55F? I can
remove them in layers some what by scraping them off, but if they are all of the same purity, just formed slower, then there is no reason for this.
    
|
|
|
semiconductive
Hazard to Others
Posts: 325
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Actually, they both increase in solubility with temperature.
NaCl goes from 357g/L to ~400g/L 0..100C
_KCl goes from 276g/L to ~575g/L 0..100C
Have you done any calculations with common ion effect taken into account, to try and figure out how much water you should leave to separate them at
higher temperatures?
Out of curiosity, the original "No-Salt" found in most stores has no NaCl in it ... and that's what I use, because re-crystallization is pretty clean.
Are you trying to separate a 95% KCl vs. 5% NaCl because of cost, or do you not have the original "No Salt" available?
The worst separation of KCl from NaCl is just above room temperature. If you can do high vacuum distillation (with a pump) near freezing / freeze
drying ... that would allow you to get the most KCl out while keeping most NaCl in Solution. The trick is to remove the moisture while keeping it
cold. ( Alternately, a cheap closed cycle high pressure CO2 could be built to bubble 45PSI CO2 through the brine, and a desiccant used to extract the
water if a high vacuum pump isn't available. )
I don't have much experience separating the two salts; but KCl is less dense than NaCl; Since they both have chloride that difference means the sodium
atom must be smaller than potassium. When it comes to being incorporated into a crystal, sodium is going to have an easier time replacing potassium
than vice-versa. The crystal lattice has to distort less, and therefore I would expect sodium impurities in bulk KCl to be easily possible; but it
would be harder for potassium to insert itself into bulk NaCl.
Another thought I have (theoretical) is that you might be able to tip common ion effect to your advantage. Right now chloride ION wants to reduce
solubility of both substances -- KCl and NaCl. But, you might be able to add cream of tartar (potassium hydrogen tartrate, 62g/L at 100C), or neutral
tartrate (P564. "Solubilities of Inorganic and Organic Compounds", Atherton Sidel: "(K2C4H4O6)2.H2O" @16 degrees, 100g water dissolves 138g neutral
tartrate ) or potassium carbonate (infinitely soluble/deliquescent);
Any of the above organic potassium salts would selectively want to remove the potassium salt from solution without really affecting the sodium
solubility. Potassium carbonate impurities crystalized in KCl can easily be gotten rid of by wetting with a tiny bit of muriatic acid (HCl). Since
K2CO3 is extremely hygroscopic ... it's not going to want to go into the KCl salt crystal in the first place, but HCl would immediately convert it to
KCl.
Since both potassium tartrates are organic, they won't fit in the KCl lattice and shouldn't embed in the crystal; But even if some does, the salt
can be burnt to oxidize the tartrate, and then add a little HCl to convert the potassium into KCl. I would expect that to be an easy solution.
[Edited on 22-1-2018 by semiconductive]
|
|
|
RogueRose
International Hazard
Posts: 1593
Registered: 16-6-2014
Member Is Offline
|
|
| Quote: Originally posted by semiconductive | Actually, they both increase in solubility with temperature.
NaCl goes from 357g/L to ~400g/L 0..100C
_KCl goes from 276g/L to ~575g/L 0..100C
Have you done any calculations with common ion effect taken into account, to try and figure out how much water you should leave to separate them at
higher temperatures?
Out of curiosity, the original "No-Salt" found in most stores has no NaCl in it ... and that's what I use, because re-crystallization is pretty clean.
Are you trying to separate a 95% KCl vs. 5% NaCl because of cost, or do you not have the original "No Salt" available?
The worst separation of KCl from NaCl is just above room temperature. If you can do high vacuum distillation (with a pump) near freezing / freeze
drying ... that would allow you to get the most KCl out while keeping most NaCl in Solution. The trick is to remove the moisture while keeping it
cold. ( Alternately, a cheap closed cycle high pressure CO2 could be built to bubble 45PSI CO2 through the brine, and a desiccant used to extract the
water if a high vacuum pump isn't available. )
I don't have much experience separating the two salts; but KCl is less dense than NaCl; Since they both have chloride that difference means the sodium
atom must be smaller than potassium. When it comes to being incorporated into a crystal, sodium is going to have an easier time replacing potassium
than vice-versa. The crystal lattice has to distort less, and therefore I would expect sodium impurities in bulk KCl to be easily possible; but it
would be harder for potassium to insert itself into bulk NaCl.
Another thought I have (theoretical) is that you might be able to tip common ion effect to your advantage. Right now chloride ION wants to reduce
solubility of both substances -- KCl and NaCl. But, you might be able to add cream of tartar (potassium hydrogen tartrate, 62g/L at 100C), or neutral
tartrate (P564. "Solubilities of Inorganic and Organic Compounds", Atherton Sidel: "(K2C4H4O6)2.H2O" @16 degrees, 100g water dissolves 138g neutral
tartrate ) or potassium carbonate (infinitely soluble/deliquescent);
Any of the above organic potassium salts would selectively want to remove the potassium salt from solution without really affecting the sodium
solubility. Potassium carbonate impurities crystalized in KCl can easily be gotten rid of by wetting with a tiny bit of muriatic acid (HCl). Since
K2CO3 is extremely hygroscopic ... it's not going to want to go into the KCl salt crystal in the first place, but HCl would immediately convert it to
KCl.
Since both potassium tartrates are organic, they won't fit in the KCl lattice and shouldn't embed in the crystal; But even if some does, the salt
can be burnt to oxidize the tartrate, and then add a little HCl to convert the potassium into KCl. I would expect that to be an easy solution.
[Edited on 22-1-2018 by semiconductive] |
The mixture is a fertilizer mix of pink muriate of potash that is extremely affordable and about the same purity as the water-softener KCl but it has
this red/pink greasy slime (it almost seems like oily rust or maybe oily clay as it is SUPER fine in consistency and is easily removed with cotton
filters - it seems magnatized to cotton - but washes clean with dish soap). I wanted to see how pure a product I could get with this material as a
starting point and how much effort is needed.
I tried putting the solution in the freezer and it eventually froze solid but a good bit of KCl dropped out of solution below O C, but it seems I left
it in too long and it froze solid, then the KCl couldn't be removed and upon thawing, it dissolved the KCl back into solution, so there is a fine line
where the temp is just cold enough to crash out KCl and not freeze the solution. I suspect that the more 95/5 mix I add to the solution, the lower
this temp will be due to a higher concentration of NaCl building up in the solution, depressing the freezing point. (so if 1 mole per liter is
dissolved per "batch", then each time there is a 5% increase in the level of NaCl in the brine mixture).
I'd really like to try the vacuum approach as this may be the most promising avenue to pursue.
I can pull greater than -700 torr (about -715 to -730 torr after a minute or so) at full vacuum and I don't have a good way of controlling the amount
of vacuum pulled so it is kind of either full on/off.
I thought I would drop the temp to at least 0 C, probably lower to ~ 15F or -10C (freezer can get to -10F or -24C) then put it under vacuum. The
thing is IDK how long is needed for the KCl to crash out, if it happens pretty quickly (which I would suspect) then I would assume that it would also
dissolve the KCl back into solution pretty quickly when the vacuum is cut off. I guess it may be possible to add a line to draw off the liquid after
the salt crashes out, at least the portion of liquid above the level of crashed salt. Do you think this is necessary or beneficial or would a quick
decantation or just pouring entire solution/crashed salt into a large nylon cloth filter, which would take about 5-10 seconds to do 1/2-1 gallon of
solution.
My biggest question is how much NaCl is going to be crystallize with the KCl.
I've been filtering every solution through activated carbon as it often has a slight yellow tint and this filtration makes it crystal clear - the
problem is that it seems to clog easily if the solution is even slightly super saturated for the room temp of the filter. I need a way to keep the
filter heated to about 10-40 degrees warmer than what it is now.
|
|
|
semiconductive
Hazard to Others
Posts: 325
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
| Quote: Originally posted by RogueRose |
I'd really like to try the vacuum approach as this may be the most promising avenue to pursue.
I can pull greater than -700 torr (about -715 to -730 torr after a minute or so) at full vacuum and I don't have a good way of controlling the amount
of vacuum pulled so it is kind of either full on/off.
I thought I would drop the temp to at least 0 C, probably lower to ~ 15F or -10C (freezer can get to -10F or -24C) then put it under vacuum. The
thing is IDK how long is needed for the KCl to crash out, if it happens pretty quickly (which I would suspect) then I would assume that it would also
dissolve the KCl back into solution pretty quickly when the vacuum is cut off.
|
I have no way of knowing how much will crash out and redissolve. My skill level with mass action laws, pressure, and water aren't high enough yet.
I'm not sure how to handle temperature at the same time as pressure when it comes to solubility.
Vacuum lines remove water from the bath at cold temperatures. So any potassium chloride that crashes out due to vaccum will NOT redissolve once
vacuum is removed. Temperature is your only real enemy. The only additional complicating issue you really have is that a thermmal gradient will
cause circulation of water due to water density changes and gravity; The water "convection" currents will prevent the crystal purity to be solely
determined by layering.
I disolved KCl with potassium bitrate to eplore the effect. I burnt it afterward to see where the bitartrate remained. It did crudely fit into
layers, but it took up a significant portion of the salt. You've got crystals with air spaces between them just like I got, so water circulation will
still happen. Slow crystallization in a thermos bottle is about the best defense possible, but it's still not going to be perfect. The object is to
keep the thermal gradient to a minimum so water circulation is minimum while evaporating water.
The issue is that when water is hot (above 4C when distilled) water expands as it gets hotter and becomes less dense. Less dense water tends to float
upward against gravity and more dense water tends to sink downward. So, when you are abolve 4C and the bottom of your fluid is warmer than the top (a
common situation on stoves or in thermos bottles with the heat leaking cap on "top") the hotter water from the bottom wants to "rise" and the colder
water on top wants to "sink"; the net result is usually that a vortex (like a tornado but very slow) will form and exchange water from the top and
bottom in a semi-circular path.
When you pull a vacuum on hot water, you make the top of the water even colder by evaportation which will increase the convection currents even more
for as long as the water is warm.
Not only is the circulation a problem, but crystals form quickest where the water is coldest. So depending on how fast the water evaporates ...
micro-crystals will want to form at the top of the thermos ... and then fall to the bottom of the flask stirring up the water as they go.
In order to combat this, you would need some kind of heater at the top of the thermos which regulates the temperature at the cap slightly hotter than
that of the thermos; say 5 degrees C. This will reverse the thermal gradient so that the water at the bottom of the flask wants to stay there because
the least dense liquid is already at the top of the flask.
Unfortunately, somewhere around 4C -- the density change of water reverses direction and begins to expand as it gets colder (closer to ice); This is a
weird anomaly of water and that number of 4C probably changes with salinity ... I don't know how much.
But basically as you drop below ~4 Co the top heater probably needs to be turned off.
The real problem is that you are doing this all by hand, and your patience and vigilance is the risk; because if you aren't paying attention at the
exact right moment, things will fail.
I could easily describe how to build an electronic circuit to regulate temperature, auto start a brine pump, shut it off, and turn the vacuum pump on
and off.... but without knowing the best end points from experience, and having no calculations yet ... I wouldn't be able to help you choose the
right circuitry for your job.
I'm familiar with physical chemistry, but I don't have enough experience with crystallization to give you a foolproof method.
I'm working on crystallizations in another thread, with potassium sulfate and copper nitrate; and the co-crystallization in that case should be easy
compared to KCl and NaCL because the two substances are very different; but they surprizingly co-crystalize into a yellow mass rather than into a
white and green mass (separately) even though the solubility of the two substances is very different.
I'm considering building an apparatus, similar to a distillation system, which step-wise heats a fluid in an insufficient amount of water to dissolve
a tiny amount of the salt and then auto transfer the solution to another flask where it is slightly cooled to cause crystalization; and then
re-circulates the cooled fluid to the heater bath. Such a system would work identically to a still .. but for solid->liquid->solid state rather
than liquid->gas->liquid state conversions.
[Edited on 28-1-2018 by semiconductive]
|
|
|
NEMO-Chemistry
International Hazard
Posts: 1559
Registered: 29-5-2016
Location: UK
Member Is Offline
Mood: No Mood
|
|
| Quote: | | Quote: Originally posted by semiconductive | | Quote: Originally posted by RogueRose |
In each of these I could easily use a silicone or rubber stopper and attach a vacuum pump to reduce the pressure. I would think that this would cool
the liquid faster, at least initially, but IDK how that would work or if there are any benefits to doing so as opposed to allowing the temp to just
fall slowly. |
|
For recrystallization, often impurity atoms change the aspect ratio of crystal growth; this is compounded with how quickly the crystals grow. The
slower a crystal grows (percentage wise), typically, the more pure and regularly shaped it will be. In crystal growing competitions, often the
hardest part is getting the initial seed crystal to be the only one and highly pure. Typically, this is achieved by controlling the super-saturation
of the solution and allowing it to drop very slowly in temperature at first. Additonally, the chamber is typically kept in the dark, very still (no
shaking) and at a very even temperature for days just to start the crystal growth. The process becomes less critical once the initial crystal is
formed.
There's a few issues with using a vacuum pump. The first is that water's energy of vaporization actually rises as the temperature drops. eg: The
energy lost per mole of water actually increases as the water temperature drops. Depending on whether your vacuum is constant pressure or constant
volume loss/minute, you may actually end up cooling the water faster as the temperature drops.
The second issue is that Silicone has around 1 micron holes in it. In general, it's a non-polar substance so water or polar molecules don't want to go
through it. However, plain old air will actually work it's way through silicone fairly quickly. So, even though rubber is less chemically inert ...
true rubber is actually a better seal than silicone for vacuum applications.
To keep a thermos bottle at an even temperature, while loosing water vapor; there is a relatively easy way to increase the insulation effect while
removing vapor. Putting two concentric tubes (Horizontal/spiraled) into the thermos bottle cap and insulating the tubes outsize the thermos allows
air to be sucked in the outer tube, while water vapor is pulled out the center tube. (You could even coil them inside a second thermos bottle
allowing the water condensate to collect in the second thermos. ) The result is that a significant portion of the heat of vaporization will be
transferred from the liquid in the center tube into the air in the outer tube. Think insulated allihn condenser ... as the water vapor condenses in
the center tube, the heat is transferred to the air in the outer jacket. The longer the allihn condensor, the higher the total heat transfer will be
and the lower the total heat loss. The only important issue is that air in the outer tube move in the opposite direction than water vapor in the
inner tube. You don't even need a vacuum pump ... but a simple aquarium air pump to pressurizing the outer tube will have a similar effect.
[Edited on 21-1-2018 by semiconductive] |
This sounds really interesting, but I am having a hard time visualizing it.
|
|
|
semiconductive
Hazard to Others
Posts: 325
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Which part? The concentric tube heat exchanger, or ??
|
|
|
|








