Complete DNA Synthesis for Viral Vector
Synthetic Viral Synthesis
Step I:Custom Oligonucleotide Synthesis
Materials
•Commerical Nucleic Acid Synthesizer
•Solution of the four DNA phosphoramidite monomers (bases)
•All the 5’-hydroxyl groups must be blocked with a DMT group for all four bases
•All phosphorus linkages must be blocked with a cyanoethyl group.
•Blocking solutions
•Reaction chamber and a type of solid support such as controlled pore glass
•The solid support should be prepared with the desired first base already attached via an ester
linkage at the 3’-hydroxyl end.
•Dichloroacetic acid or trichloroacetic acid
•Tetrazole
•Acetic anhydride and N-methylimidazole
•Dilute iodine in a water/pyridine/tetrahydrofuran solution
•Concentrated ammonia hydroxide.
•Materials for one desalting method
Process
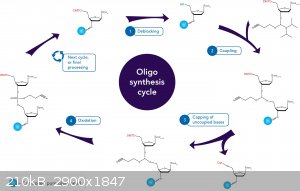
Step A: De-blocking
The first base, which is attached to the solid support, is at first inactive because all the active sites
have been blocked or protected. To add the next base, the DMT group protecting the 5'-hydroxyl
group must be removed. This is done by adding a base, either dichloroacetic acid (DCA) or
trichloroacetic acid in dichloromethane (DCM), to the reaction column. The 5’-hydroxyl group is now the only reactive group on the base monomer.
This ensures that the
addition of the next base will only bind to that site. The reaction column is then washed to remove
any extra acid and by-products.
Step B: Base Condensation
The next base monomer cannot be added until it has been activated. This is achieved by adding
tetrazole to the base. Tetrazole cleaves off one of the groups protecting the phosphorus linkage.
This base is then added to the reaction column. The active 5’-hydroxyl group of the preceeding
base and the newly activated phosphorus bind to loosely join the two bases together. This forms
an unstable phosphite linkage. The reaction column is then washed to remove any extra
tetrazole, unbound base and by-products.
Step C: Capping
When the activated base is added to the reaction column some does not bind to the active 5’-
hydroxyl site of the previous base. If this group is left unreacted in a step it is possible for it to
react in later additions of different bases. This would result in an oligonucleotide with a deletion.
To prevent this from occurring, the unbound, active 5’-hydroxyl group is capped with a protective
group which subsequently prohibits that strand from growing again. This is done by adding acetic
anhydride and N-methylimidazole to the reaction column. These compounds only react with the
5’-hydroxyl group. The base is capped by undergoing acetylation. The reaction column is then
washed to remove any extra acetic anhydride or N-methylimidazole.
Step D: Oxidation
In step 2 the next desired base was added to the previous base, which resulted in a unstable
phosphite linkage. To stabalize this linkage a solution of dilute iodine in water, pyridine, and
tetrahydrofuran is added to the reaction column. The unstable phosphite linkage is oxidized to
form a much more stable phosphate linkage.
Repeat as need based on length desired between 1 and 10,000 times.
Final Product: DNA Chains from 1 to 10,000 base pairs in length.
Step II:Molecular Cloning: Polymerase Chain Reaction(PCR)
Materials
-Thermal Cycler
-Taq polymerase
-Generated DNA Fragments
Process
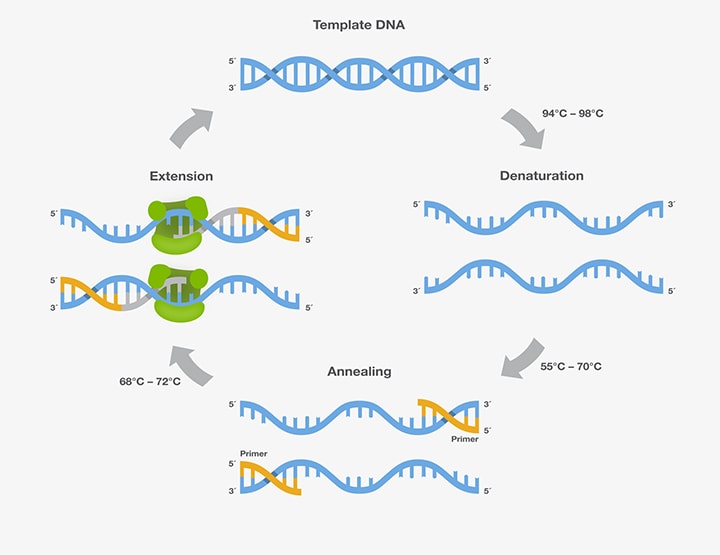
A. Denaturation - the DNA is heated usually to 95C to render it single-stranded
B. Annealing - the two primers bind the appropriate complementary strand; the temperature for
this step varies depending on the of size of the primer and its homology to the target DNA
C. Primer Extension - DNA polymerase extends the primer by its polymerase activity; this is
done at a temperature optimal for the particular polymerase that is used; currently the most
popular enzyme for this step is Taq polymerase, the DNA polymerase from the thermophilic
("heat-loving) bacteria Thermus aquaticus; the extension is performed at 72C. These steps are
repeated from 28-35 times. Since the reaction is essentially exponential and since each cycle is
about 5 minutes, a large quantity of DNA can be produced for analysis in as little as several
hours.

Final Product: Exponential DNA cloning
Step III:Mass Ligation Reaction or Standard Ligation Reaction
Materials
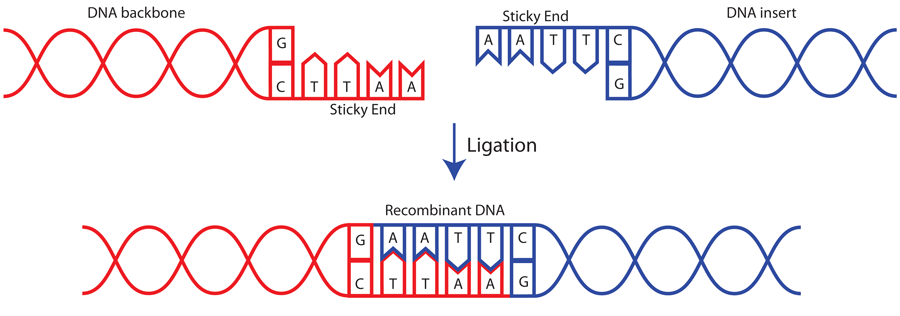
•Two or more fragments of DNA that have either blunt or compatible cohesive ("sticky") ends.
•A buffer which contains ATP. The buffer is usually provided or prepared as a 10X concentrate
which, after dilution, yields an ATP concentration of roughly 0.25 to 1 mM. Most restriction
enzyme buffers will work if supplemented with ATP.
•T4 DNA ligase. A typical reaction for inserting a fragment into a plasmid vector (subcloning)
would utilize about 0.01 (sticky ends) to 1 (blunt ends) units of ligase.
Process
The optimal incubation temperature for T4 DNA ligase is 16C and when very high efficiency
ligation is desired (e.g. making libraries) this temperature is recommended. However, ligase is
active at a broad range of temperatures, and for routine purposes such as subcloning,
convenience often dictates incubation time and temperature - ligations performed at 4C overnight
or at room temperature for 30 minutes to a couple of hours usually work well.
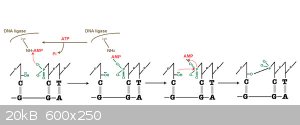
Final Product: Every possible recombination or a Single Recombination
Step IV:Activation and Reproduction : Plasmid Vector
(sub-cloning)
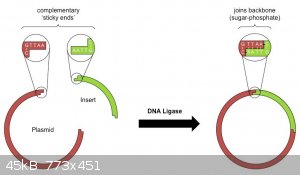
Materials
-Anything with ribosomes
-NotI Ligase
Process
Same as step III
Final Product: Viral Vector
|








