Microtek
National Hazard

Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
Lab scale contact process for H2SO4 (report)
(I realize this has been discussed before, but not recently AFAIK, so I hope I'm not breaking any rules)
I would like to report success at the contact process for sulfuric acid.
In (some of?) the EU, sulfuric acid above 50 % concentration has been banned as part of an anti-terrorism package. While it is possible to buy such
(50%) acid, and concentrate it by boiling it down, it is difficult to achieve very highly concentrated acid since decomposition/evaporation losses get
more significant the higher the acid concentration becomes. Therefore, it would be practical to be able to fortify existing acid up to (or beyond) 100
%. To achieve this I have experimented with a lab scale adaptation of the contact process. For this stage of the experiments, I decided to avoid the
additional complication of a sulfur burner and instead generate the SO2 from a wet process.
SO2 from the reaction of sodium metabisulfite with hydrochloric acid was mixed with air from an aquarium pump and dried in a silica gel column (fig
1). Indicating silica gel is very practical here, since you can easily see when it is time to refresh it.
See attached image 1: Production and drying of SO2
The dry gas stream was fed into a quartz tube furnace containing catalyst on silica gel (kitty litter).
See attached image 2: Furnace inlet
The catalyst was prepared by dissolving vanadium pentoxide in oxalic acid, forming a clear blue solution. This solution was then absorbed in (and
adsorbed on) kitty litter silica gel, and dried. Afterwards, it was heated to ca. 200 C to decompose the oxalate and leave V2O5. A few centimeters of
this catalyst were placed into the furnace and held in place with ceramic wool (aluminum silicate). The furnace was heated to ca. 450 C by means of a
nichrome element and an SCR. I had no active temperature control.
The stoppers for the furnace are made of PTFE which I turned on my lathe. A tight seal was ensured with PTFE plumbers tape.
From the furnace, the gas stream was led into an absorption tower consisting of two straight line vacuum adaptors and a Graham condenser
See attached image 3: Absorber column
I would have liked my absorber to be a packed column, but this was all I had access to at the time.
Concentrated sulfuric acid (144.4 g, of nominally 96%) was run through the tower from the pressure equalizing dropping funnel at a low enough rate
that it formed a stream at the “bottom” of the windings of the coil without blocking gas flow. This corresponded to about one drop per second.
When all of the sulfuric acid had run through, the bottom 250 ml RBF was removed, stoppered and weighed to monitor weight gain.
Results
The RBF including acid, stopper and cork ring was weighed before the first run, and after each run thereafter:
Run 1: 15.8 g metabisulfite, 7.4 g mass gain, 55.6% SO3 yield
Run 2: 62.1 g metabisulfite, 28.9 g mass gain, 55.3% SO3 yield
Run 3: 37.3 g metabisulfite, 22.1 g mass gain, 70.4% SO3 yield
“Metabisulfite” notes how much of this reagent the SO2 generator contained at the beginning of the run. A slight excess of aqueous hydrochloric
acid was added through the dropping funnel mounted on the three-necked flask. The last neck was fitted with a thermometer adaptor for the tube from
the aquarium pump. This way, the liberated SO2 was admixed with the entering air and carried through the system.
The yield calculation is based on each metabisulfite ion liberating 2 molecules of SO2, and is the overall, effective yield, assuming that all mass
gain is due to SO3 absorption.
The hoses I used were PVC from the generator to the furnace, PTFE from the furnace to the absorber (except for a small piece to connect the PTFE to
the glass vacuum adaptor), and PVC again from the top of the absorber to the exhaust bubbler. Note that where SO3 had come in contact with anything
other than glass or PTFE, extensive charring was the result. The charring of the PVC hose before the furnace was due to an experimental run with pure
oxygen (from electrolysis) as the carrier gas. In this run, the flow rate was too low, and quite a bit of back flow occurred. Likewise, the charring
after the absorber was primarily due to another experiment with a simple gas washing bottle for absorbing the SO3 into the concentrated sulfuric acid.
Doing it this way is completely ineffective, and practically all of the product was lost as acid mist in the exhaust bubbler.
The exhaust bubbler itself is there primarily as a means to monitor the process. Flow rate can be judged by the bubbles, while absorption efficiency
can be seen from how much acid mist is produced.
When the process is begun, but the flow of sulfuric acid has not yet been turned on, an impressively thick, white fume, much like stage smoke (dry ice
in water), is produced at the exhaust bubbler.
Analysis
The product was observed to be fuming, much like highly concentrated nitric acid.
A small amount was transferred to a small pre-weighed flask and weighed precisely (1.660 g). It was then slowly diluted with demineralized water (to
ca. 20 ml) and titrated with dilute NaOH solution which had been calibrated with analytical potassium hydrogen phthalate as primary standard.
The sample titrated as 102.2 % sulfuric acid, corresponding to 9.9 % free SO3.
  
[Edited on 6-2-2017 by Microtek]
[Edited on 6-2-2017 by Microtek]
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Nice work!
Indicating silica gel: Is this something you made or is it commercially available?Nvermind, I see it's available at Walmart.  . Nice use of otc materials. . Nice use of otc materials.
[Edited on 6-2-2017 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Awesome work Microtek !
It gladdens my heart to see Real-World Chemistry return to this forum.
|
|
|
j_sum1
Administrator
Posts: 6320
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
First glance, this looks stunning.
I am going to have to sit down and read this properly.
It is a good trend when there are viable home lab procedures being developed for sulfuric acid and nitric acid at the same time.
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
This is impressive. I am definitely going to pick up some vanadium pentoxide at my earliest opportunity.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Nice reduction to practice. I'm sure with further time and resources you can get it to run more efficiently.
I have a question about the catalyst... Usually the supported catalyst is prepared from ammonium vanadate - what made you decide to use oxalic acid to
dissolve your vandium pentoxide? Does it provide advantage over using ammonia? Can you provide a more detailed procedure for the preparation? Also,
have you considered adding potassium sulfate as a promotor? From what I recall, it frovides a liquid catalyst phase on the support (at operating
temperatures) which provides better conversion/throughput.
Edit: Actually, are you sure its V2O5 formed on the support after calcination? I recall oxalic acid can reduce some metal species, particularly those
in the higher oxidation states. I suspect the vanadium is not +5 in the solution, given the blue colouration, but whether it returns on activation at
temperature or not I don't know?
[Edited on 6-2-2017 by DJF90]
|
|
|
Amos
International Hazard
Posts: 1406
Registered: 25-3-2014
Location: Yes
Member Is Offline
Mood: No
|
|
Quote: Originally posted by DJF90  |
Edit: Actually, are you sure its V2O5 formed on the support after calcination? I recall oxalic acid can reduce some metal species, particularly those
in the higher oxidation states. I suspect the vanadium is not +5 in the solution, given the blue colouration, but whether it returns on activation at
temperature or not I don't know?
[Edited on 6-2-2017 by DJF90] |
I can't give you an exact temperature, but I can confirm that it reverts back to vanadium pentoxide with strong heating; annoyingly so. I was
attempting to isolate the vanadyl compound in solid form but left the solution on the hot plate too long and came back to yellow-orange vanadium
pentoxide that I have since recycled for other vanadium compounds.
|
|
|
Chemetix
Hazard to Others
Posts: 375
Registered: 23-9-2016
Location: Oztrayleeyah
Member Is Offline
Mood: Wavering between lucidity and madness
|
|
Awesome! I have been wanting to try this exact technique, you've done a nice job. Was going to try oxidising SO2 with NO2 first.
|
|
|
Microtek
National Hazard
Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
First of all: Thanks for the kind words everyone!
@ DJF90:
I tried using ammonium metavanadate at first, but couldn't get the V2O5 to dissolve in aqueous ammonia. I thought, maybe I wasn't using a high enough
concentration (I found, by titration, that my ammonia solution had dropped to ca. 21% over the years). However, when I researched preparation
procedures of ammonium metavanadate, I discovered that it is actually quite insoluble. I wanted a soluble vanadium salt that could be
deposited on a suitable support and then be turned back into V2O5. Vanadyl oxalate was what my research turned up.
It is prepared by an adaptation of the process in US3689515: Three equivalents of oxalic acid dissolved in water is heated to ca. 90 C, and one
equivalent of vanadium pentoxide is added with stirring. The solution turns an impressive peacock blue as the reaction proceeds. I then added
potassium sulfate to act as a promoter, since I have also read that this is advantageous.
After cooling, I added some of the solution to a spoonful of kitty litter zeolite. I used enough solution that not all of it was absorbed, since I
wanted to be certain that the outer surface was coated as well (since I am not certain that much of the reaction will take place on the inside
surface). I swirled the beaker intermittently as the excess liquid evaporated to leave the catalyst. After air drying, I heated it in the oven at ca.
200 C, which gave a colour change of the (outer) surface to orange/red.
I figured that, since the catalytic process is probably a cycle of oxidation and reduction, it probably wouldn't be too sensitive as to the exact
oxidation state of the vanadium.
Pictured below is a comparison of the different stages of the catalyst preparation:
Sample at the left is untreated kitty litter zeolite. Top mid is after air drying of the vanadyl oxalate impregnated zeolite, and right is after oven
drying. Bottom mid is after calcining for about 30 seconds at red heat (blowtorch). Note that the colouring of the final product only develops after
standing for a few minutes; immediately following the calcining, it is almost colourless.
![WP_20170207_005[158].jpg - 349kB](https://www.sciencemadness.org/whisper/files.php?pid=473967&aid=56858)
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
@Microtek - Thanks for taking the time to elaborate. I took a few minutes after my last comment to have a look into the dissolution of vanadium
pentoxide in oxalic acid and found (as you mention) that vanadyl oxalate is formed. I agree with you that oxidation state is unlikely to be critical
due to the redox-cycling of the catalyst under operating conditions. One thing that may improve efficiency is to heat in a stream of air prior to
generation of the sulfur dioxide, just to ensure it is completely oxidised before the catalytic cycle commences.
I think a packed absorption tower at this stage may be excessive, as your improvised apparatus appears to do a good job of absorbing the sulfur
trioxide (I'm sure you'd know if it wasn't). Perhaps if you scale up for higher throughput then it will become a necessary factor. One thing you
haven't mentioned in your initial report was the catalyst bed dimensions/mass of catalyst/estimated loading (%w/w V2O5 on support?).
|
|
|
wg48
National Hazard
Posts: 821
Registered: 21-11-2015
Member Is Offline
Mood: No Mood
|
|
Microtek;
Buying silica gel as kitty litter may be cheap source of silica gel, but apparently some silica gel types of kitty litter have additives such as
scent and indicator and some may be a clay type of material. The brand you used does look like silica gel and I assume it contains no additives. So
which brand of kitty litter did you use?
|
|
|
Microtek
National Hazard
Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
@ wg48:
It is called "Fresh&Easy" and says that it contains an "acidity regulator" in addition to silicate, but also boasts of an "anti-bacterial shield".
In truth, Idon't think it is extremely important, since one of the reasons that V2O5 is a preferred catalyst in industry, is that it is not easily
poisoned (unlike platinum).
I was wondering how much SO2 might have contaminated my product since, (assuming a less than 100% conversion of the SO2) I am contacting the
concentrated sulfuric acid with both SO2 and SO3 in the absorber. To answer that question, I removed a sample of my 10% oleum (0.456 g) and slowly
diluted it with demineralized water (by diffusion initially) up to ca. 20 ml. It was then titrated with KMnO4 solution of 0.0000525 M concentration.
The KMnO4 was merely weighed off and dissolved since I have no primary standard for it. However, if some of the KMnO4 is actually MnO2, the titration
will overestimate the content of reducing substances due to the ratio of molar masses to stoichiometric coefficients.
The end point of the titration was reached after 0.2 ml, corresponding to a max content of 0.037 % SO2 (or 0.042 % HCl). It is also entirely possible
that carbonized residues (the brown colour of the product) was what was oxidized in the titration.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Here in Spain there appear to be just two types : silica or bentonite clay.
The cheaper the cat litter, the better.
Those additives cost money to add !
|
|
|
ecos
Hazard to Others
Posts: 464
Registered: 6-3-2014
Member Is Offline
Mood: Learning !
|
|
where did u get sodium metabisulfite from?
|
|
|
Geocachmaster
Hazard to Others
Posts: 146
Registered: 5-3-2016
Location: Maine, USA
Member Is Offline
Mood: Corroded, just like my spatulas
|
|
The most OTC source of sodium metabisulfite is likely from stump remover. Bonide sells a stump out which is listed as 99% metabisulfite on the sds.
Some stump removers are potassium nitrate as well, so check the MSDS. Although both nitrate and metabisulfite are useful. Other than that look on eBay
or some other website, plenty of chemicals can be found online.

I got 450g (one pound) for about USD$8 from a local hardware store.
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
Brewing stores sell it for sterilization and some process used for making wines. It can also be used to remove unwanted ketones from alcohols.
|
|
|
PirateDocBrown
National Hazard
Posts: 570
Registered: 27-11-2016
Location: Minnesota
Member Is Offline
Mood: No Mood
|
|
I found some sodium metabisulfate the other day as carpet stain remover. It had some surfactants in it, but if all it's used for is generating SO2,
that won't be a problem. (If I use it for cleaning, it's likely even better!)
I have found it in photo shops, Kodak still makes bags of it.
I have also found it in a brewing supply store.
It's actually one of the easier chemicals to get.
https://www.janilink.com/product_info.php?products_id=14944
[Edited on 2/9/17 by PirateDocBrown]
|
|
|
Microtek
National Hazard
Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I got it from a boating store, but have also seen it in shops that cater to the needs of wine-making enthusiasts. I'm in Scandinavia though, so what's
available to me is often not what's available to Americans. Having said that, the use of metabisulfite was simply a way to deal with one design
problem at a time; designing a reliable sulfur burner is not trivial, in my experience. But now that I have managed to get a reasonably reliable
method of turning SO2 into oleum, the next step will be reliably turning sulfur into SO2. I'm not as ambitious as Axehandle was in his endeavours some
years ago; I will settle for a batch process that is able to handle maybe 100g of sulfur per batch. I think I will probably cheat, and use an oxygen
enriched air stream, since that does make for a trivial design problem.
[Edited on 9-2-2017 by Microtek]
|
|
|
ecos
Hazard to Others
Posts: 464
Registered: 6-3-2014
Member Is Offline
Mood: Learning !
|
|
sounds good. i will try to find sodium metabisulfate 
@Microtek: to go from sulfur to sulfuric acid, i think you would need vanadium(V) oxide as catalyst.
hope this is easy to find
|
|
|
Microtek
National Hazard
Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
That is what I have been using (vanadium(V)oxide is the same as V2O5).
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
@ PirateDocBrown and ecos,
METABISULFITE not METABISULFATE...
[Edited on 14-2-2017 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Belowzero
Hazard to Others
Posts: 173
Registered: 6-5-2020
Location: Member Is Offline
Member Is Offline
|
|
I realize this thread is more than 3 years old but might be more relevant than ever before.
Last few days I have been reading up on the process while thinking it might be out of reach for backyarders, until I found this conversation.
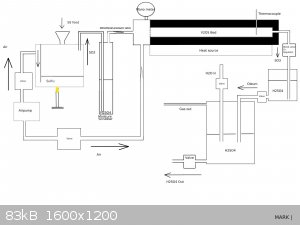
@Microtek I am curious if you ever followed up on your experiments and if you would be so kind as to draw your reactor.
I am planning to investigate a viable way to use pressure to shift the equilibrium of SO2 + O2 -> SO3
According to the sources I have found this can be shifted with as little as <2 Bar of pressure which even regular glassware might tolerate.
The engineering part is probably the biggest challenge.
This source has a very easy to read summary of the process:
https://www.chemguide.co.uk/physical/equilibria/contact.html
*Edit
To get the juices flowing I drew up a possible reactor, please let me know what you think.
I don't want to hijack this thread , if the mods prefer a new thread please say so and I'll open up a new one.
[Edited on 6-8-2020 by Belowzero]
|
|
|
xxxhibition
Harmless
Posts: 12
Registered: 25-1-2021
Member Is Offline
|
|
Found a relativ simple looking Build up allmost Glasware here...
Sulphur trioxide - the sulphuric acid contact process
Its not cheap there but you should be able to re - build this
SO3 and Contact Process
Attachment: p3110400e.pdf (224kB)
This file has been downloaded 528 times
Attachment: 11399_01.pdf (369kB)
This file has been downloaded 511 times
[Edited on 29-1-2021 by xxxhibition]
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
| Quote: Originally posted by Belowzero |
I am planning to investigate a viable way to use pressure to shift the equilibrium of SO2 + O2 -> SO3
According to the sources I have found this can be shifted with as little as <2 Bar of pressure which even regular glassware might tolerate.
|
The equilibrium strongly favors SO3 at any temperature that "regular" (borosilicate, hopefully!) glassware can tolerate. There's no need to
mess with the pressure in those cases.
Trying to reach the temperature at which the reaction proceeds thermally is a fool's errand; materials obtainable for the amateur will not tolerate
the rxn conditions (if anything will). Just use V2O5.
|
|
|
Texium
|
Thread Moved
29-11-2023 at 19:49 |








