| Pages:
1
2 |
Furch
Hazard to Others

Posts: 130
Registered: 8-10-2006
Member Is Offline
Mood: No Mood
|
|
Potential route to amphetamine...
Howdy!
I've had this idea for a route to amphetamine for quite some time now, but I haven't bothered exploring it further, for some reason... Now, however,
I've decided to at least bring it up for discussion.
I haven't seen this route around so far, possibly because it's a no-go... I'm not claiming this is revolutionary or a great way, I just hope it will
be a working way... Anyhow, here's my idea:
Ph-CHO + CH3NO2 --Base--> Ph-CH=CH-NO2
This condensation I don't even need to explain, as it's such a well known reaction in these circuits.
Ph-CH=CH-NO2 --NaBH4--> Ph-CH2-CH2-NO2
Lots of sources for this kind of reduction as well...
Ph-CH2-CH2-NO2 --1) Base 2) CH3I--> Ph-CH2-CH(CH3)-NO2
Here's where things get unexplored, I think. However, I don't see any reason for this textbook example not to work. However I haven't got any
references or anything... Standard alkylation of nitro enolate.
Ph-CH2-CH(CH3)-NO2 --Zn/HCl/other-reducing-system--> Ph-CH2-CH(CH3)-NH2
This is also a very familiar reduction which I don't think needs further explanation.
Reaction sequences with names:
Benzaldehyde ----> beta-nitrostyrene ----> 2-phenylnitroethane ----> 1-phenyl-i-nitropropane ---->
1-phenyl-i-propylamine (amphetamine)
Sorry for not supplying graphic reaction mechanisms, I don't have access to that sort of program at the moment.
Any criticism is welcome!
- Furch
|
|
|
Chris The Great
Hazard to Others
Posts: 463
Registered: 29-10-2004
Location: Canada
Member Is Offline
Mood: No Mood
|
|
Interesting, I've never head of the alkylation of the nitro compound before. Do you have any examples or information on the reaction? I've never
even heard of it, but if it is a standard reaction I see no reason why it wouldn't work as presented.
|
|
|
solo
International Hazard
Posts: 3975
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
Your proposed synthesis looks much like this ......but more involved .....solo
----------------------------------------------------------------------
The phenylnitropropene route
Condensation of benzaldehyde with nitroethane yields l-phenyl-2-nitro-propene. Hydrogenation of the double bond and reduction of the nitro-group give
the amphetamine.

in which (1): 20- 100°C. (2): 1-80 Atm.
(3): LiAlH 4, H 2 and Raney-Ni or Pd/C.
(4): CH 3OH; C 2H 5OH; H 20/HCOOH; CH 3COOH/C 2H 5OH.
Reaction conditions vary widely [25, 26] and electrolytical reduction has been mentioned [ 27] . This synthesis has been of importance in Sweden in
illegal amphetamine production [ 28] .
We have given a rather extensive description of some methods for the production of illegal amphetamine for which the starting material can be
purchased easily and in which the chemical operations are not too complicated. Many other synthetic routes for amphetamine, often with obscure or
intricate chemicals, are known, 1but in this context are of lesser importance.
.................source,
http://www.unodc.org/unodc/bulletin/bulletin_1981-01-01_3_pa...
[Edited on 8-12-2006 by solo]
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
Organikum
resurrected
Posts: 2337
Registered: 12-10-2002
Location: Europe
Member Is Offline
Mood: frustrated
|
|
Well actually the trick is that he uses nitromethane whats not watched instead of nitrethane what is.
Sounds interesting.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Furch
Ph-CH2-CH2-NO2 --1) Base 2) CH3I--> Ph-CH2-CH(CH3)-NO2
Here's where things get unexplored, I think. However, I don't see any reason for this textbook example not to work. However I haven't got any
references or anything... Standard alkylation of nitro enolate.
|
Sorry to disappoint you, but the nitronates get O-alkylated and then the instable alkyl nitronate products rearrange to the appropriate oxime and
aldehyde/ketone. Generally there is either no C-alkylation product or it represent a very minor product. This was repeated ad nauseam at the old Hive
and still some never learned. There are only a couple of cases of C-alkylation of nitroalkanes in the literature and all are mechanistically
exceptional due to electrophile and/or nucleophile chemical properties (your substrate and MeI do not fit any such exception).
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Furch
Hazard to Others
Posts: 130
Registered: 8-10-2006
Member Is Offline
Mood: No Mood
|
|
Nicodem, I suspected you would bring me some bad news 
Anyhow... I don't mean to argue with you, but I need you to sort this out for me, if you will.
"As the character of a given reaction changes from SN1-like to SN2-like, an ambident nucleophile becomes more likely to attack with its less
electronegative atom. Therefore, changing from SN1 to SN2 conditions should favor C attack by CN-, N attack by NO2-, C attack by enolate or phenoxide
ions, and so on."
"All negatively charged nucleophiles must of course have a positive counterion. If this ion is Ag+ (or some other ion that specifically helps in
removing the leaving group), rather than the more usual Na+ or K+, then the transition state is more SN1 like. Therefore the use of Ag+ promotes
attack at the more electronegative atom."
"In many cases, the solvent influences the position of attack. The freer the nucleophile, the more likely it is to attack with its more
electronegative atom, but the more this atom is encumbered by either solvent molecules or positive counterions, the more likely is attack by the less
electronegative atom. In protic solvents, the more electronegative atom is better solvated by hydrogen bounds than the less electronegative atom. In
polar aprotic solvents, neither atom of the nucleophile is greatly solvated, but these solvents are very effective in solvating cations. Thus in a
polar aprotic solvent the more electronegative end of the nucleophile is freer from entanglement by both the solvent and the cation, so that a
change from a protic to a polar aprotic solvent often increases the extent of attack by the more electronegative atom."
These quotes are from March's advanced organic chemistry, 5th edition.
In accordance with the above given statements, C-alkylation of the nitroalkyl enolate should be favored if some reaction conditions and
reactants are carefully adjusted, such as:
* The use of alkali hydroxide as base for enolate formation, giving Li+, Na+ or K+ as counterion.
* Use of a protic solvent, such as simple alcohols (Me- Et- or PrOH/i-PrOH).
You are more than welcome to comment my above reasoning.
I don't mean to be a pain in the ass, it's just that I have a hard time swallowing that C-alkylation of nitro enolates are rarely seen... As I've seen
it lots throughout my organic chemistry "career" 
An alternative to the use of MeI as alkylating agent would then be the use of formaldehyde... This would probably give the intermediate alcohol, which
hopefully could be dehydrated to the nitropropene by phtalic anhydride (as seen in similar dehydrations), then one could take it from there.
Sincerely,
Furch
[Edited on 8-12-2006 by Furch]
|
|
|
haribo
Harmless
Posts: 24
Registered: 28-11-2006
Member Is Offline
Mood: No Mood
|
|
If you make PEA and form an imine with methyl amine, CH3I will add to that. Someone on the Hive called Labrat actually made it work. Of course, there
IS a 1 step reduction from phenyl alanine to amphetamine. It uses some pretty wacky reducing agents, thought. Tris trifluoro phenyl phosphine is the
catalyst. If I remember rightly, the reducing agent is some relative to DIBAL.
You could make alpha methyl tryptamine the same way, I assume.
|
|
|
XxDaTxX
Hazard to Self
Posts: 66
Registered: 12-3-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by haribo
If you make PEA and form an imine with methyl amine, CH3I will add to that. Someone on the Hive called Labrat actually made it work. Of course, there
IS a 1 step reduction from phenyl alanine to amphetamine. It uses some pretty wacky reducing agents, thought. Tris trifluoro phenyl phosphine is the
catalyst. If I remember rightly, the reducing agent is some relative to DIBAL.
You could make alpha methyl tryptamine the same way, I assume. |
PEA imine? Now that's really interesting. Can you draw that? =P
...Maybe you meant phenylacetaldehyde to adlimine. Then methyl grignard.
As for the original post. Its a very good reaction, and it does work =] .................................and work. =[
You have half of it up there. If you were to form the nitronate and could get reaction conditions to favor methylation on the base-deprotonated alpha
carbon, then you would get methylation on the alpha carbon ........... and base deprotonated alpha carbon, then you would get methylation on the alpha
carbon. Heheheh.
|
|
|
Furch
Hazard to Others
Posts: 130
Registered: 8-10-2006
Member Is Offline
Mood: No Mood
|
|
This reminds me, IF this idea would work out, i.e. reacting 2-phenylnitroethane with CH3I to form 1-phenyl-i-nitropropane, then one could
employ this procedure to make nitroethane from nitromethane and CH3I... Which renders my amphetamine procedure totally useless from a profit-point of
view, since solo's suggestion above can be employed in its stead (PhCHO+ EtNO2 -red-> amph.).
However I didn't come up with the idea for the sake of manufacturing amphetamine, but for the experimental value of the route.
- Furch
[Edited on 8-12-2006 by Furch]
[Edited on 8-12-2006 by Furch]
|
|
|
XxDaTxX
Hazard to Self
Posts: 66
Registered: 12-3-2006
Member Is Offline
Mood: No Mood
|
|
From your language I don't think you got my last post. Otherwise you would know your proposed step (base catalyzed alpha methylation) wont give you
what you want. Using the right solvent, you can get it to work all too well .... methylation on the alpha carbon causes the remaining alpha hydrogen
to be even more acidic than the first, resulting in dimethylation. You will yield a mixture of dimethylated and methylated products, the prior being
predominant.
[Edited on 9-12-2006 by XxDaTxX]
|
|
|
Organikum
resurrected
Posts: 2337
Registered: 12-10-2002
Location: Europe
Member Is Offline
Mood: frustrated
|
|
The direct reductionof phenylalanine to amphetamine is done with triethylsilane in trifluoroacetic acid IIRC. The silane being meanwhile a common
reagent AFAIK.
Reductions with this sytem almost always result in practically quantitative yields, thus beating the holy shit out of LiAlH.
[Edited on 9-12-2006 by Organikum]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
For the last time, (most) nitroalkanes get O-alkylated under any condition for nucleophilic substitution! There is no balancing between C- and
O-alkylation for better yield like in the case of ketone derived enolates in dependence on solvents, counterions etc. The nitronate anion (aci- form)
is about a hundred thousand times more stable that the apropriate alpha-deprotonated nitroalkanes (just check the pKa's of both forms!).
XxDaTxX, please stop spreading misinformation of the type: "...and base deprotonated alpha carbon, then you would get methylation on the alpha carbon.
Heheheh." Use references when you claim anything that goes beyond common chemical knowledge!
Only in the cases of highly and fully reversible reactions, like with the carbonyl compounds, is the O-alkylation of nitroalkanes irrelevant and one
only obtains the thermodynamic, that is, C-alylation product. Therefore, the condensation with formaldehyde is a viable option even though it is such
a waste of time for such a relatively easy molecule like amphetamine.
To put this in a graphical way, see this excerpt from the chapter "5. Formation of carbon-carbon bonds: the use of stabilized carboanions and related
nucleophiles" of the book "Guidebook to Organic Synthesis" (3rd ed.; R. K. Mackie, D. M. Smith and R. A. Aitken):
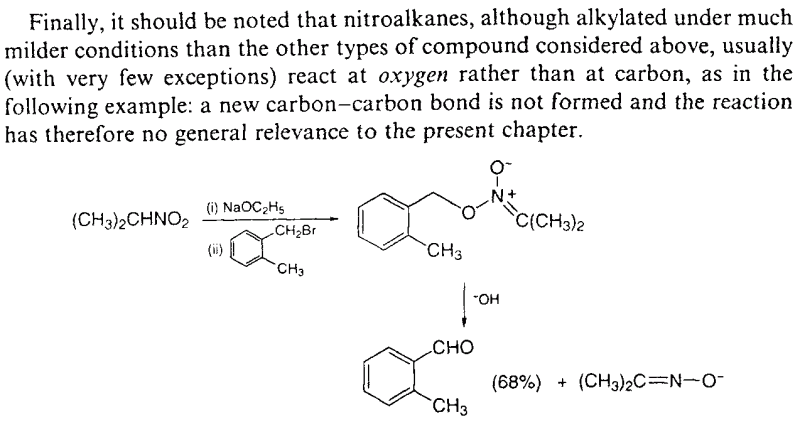
The only use of a nitroalkane alkylation with alkyl halides in the synthesis of an amphetamine is described in (attached):
Hoover F.W., Hass H.B. Synthesis of paredrine and related compounds. J. Org. Chem., 12 (1947) 501-505.
In this paper and the ones referenced in it there is a short mechanistic explanation on why it works with their electrophile and why it does not work
otherwise.
[Edited on 9-12-2006 by Nicodem]
Attachment: Synthesis of paredrine and related compounds.pdf (360kB)
This file has been downloaded 1627 times
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
XxDaTxX
Hazard to Self
Posts: 66
Registered: 12-3-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Nicodem
For the last time, (most) nitroalkanes get O-alkylated under any condition for nucleophilic substitution! There is no balancing between C- and
O-alkylation for better yield like in the case of ketone derived enolates in dependence on solvents, counterions etc. The nitronate anion (aci- form)
is about a hundred thousand times more stable that the apropriate alpha-deprotonated nitroalkanes (just check the pKa's of both forms!).
XxDaTxX, please stop spreading misinformation of the type: "...and base deprotonated alpha carbon, then you would get methylation on the alpha carbon.
Heheheh." Use references when you claim anything that goes beyond common chemical knowledge!
Only in the cases of highly and fully reversible reactions, like with the carbonyl compounds, is the O-alkylation of nitroalkanes irrelevant and one
only obtains the thermodynamic, that is, C-alylation product. Therefore, the condensation with formaldehyde is a viable option even though it is such
a waste of time for such a relatively easy molecule like amphetamine. |
All in good fun. =]
You need to CHILL ..... no really ... CHILL!
If you want the nitty gritty here you go:
The prep was a cyclization involving sulfone addition to a nitroalkene. Sulfone addition yielded bromoallyl sulfone and subsequent ring closure,
employing HMPA @ -78oC, gave . Yield was 60%, but where else are you going to get yields that high for c-alkylation to nitronate. You look it up if
you are so interested. Only reason I remember it is cause I said
"HEY! NOW I CAN TELL PEOPLE I C-ALKYLATED ON A NITRONATE IN ACCEPTABLE YIELDS!"
=P
[Edited on 10-12-2006 by XxDaTxX]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by XxDaTxX
"HEY! NOW I CAN TELL PEOPLE I C-ALKYLATED ON A NITRONATE IN ACCEPTABLE YIELDS!" |
The only nitroalkanes that get C-alkylated regardless of the electrophile nature are the ones that have an extra group with -M effect at the alpha
position to stabilize the carboanionic form. Therefore compounds of the type R-CH2-NO2 where R = -COOR, -SO2R, -NO2, -CN etc. If you have a reference
that says otherwise, please post it. Otherwise please do your best not to misinform with such statements.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
XxDaTxX
Hazard to Self
Posts: 66
Registered: 12-3-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Pure and applied Chemistry 72:1671
SULFONE ADDITIONS TO NITROALKENES
We also examined the possibility of achieving cyclopentanation with 1 and alpha,beta-unsaturated nitro compounds. Since nitro groups can readily be
removed or transformed into a keto function, this would represent a useful entry into various substituted cyclopentanes. Such a sequence would require
that the Michael addition of the sulfone carbanion be followed in tandem by a C-alkylation of the resulting nitronate ion. However, it has been shown
that nitronate anions do not undergo C-alkylation, even wit allyl halides; at best they undergo O-alkylation.
When we carried out the reaction of the lithio derivative of 1 with several conjugated nitro olefins at –100 °C, we found that syn and anti
open-chain addition products had formed with little stereoselectivity. However, in the presence of HMPA, already at –78 °C and more readily at
–40 °C, cyclization via C-alkylation of the nitronate was achieved, and the nitro substituted methylenecyclopentanes could be obtained in up to 50%
yield [9]. This represents the first examples of C-alkylation of nitronate anions (Scheme 10). In this manner 1-nitrocyclohexene can be converted into
a bicyclic methylenecyclopentane.
|
Chill
[Edited on 10-12-2006 by XxDaTxX]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Thanks for the reference. I guess it is never too late.
The example is interesting even though it bears no significance to the alkylation proposed by Furch. Apparently the nitronate gets intramolecularly
C-alkylated when the only other options is to either intramolecularly form a 7 membered ring by O-alkylation or O-alkylate intermolecularly. It is an
information that can come useful in planning the synthesis of 5-membered nitrocycloalkanes. It might even work in forming 6-membered rings.
PS: I never chill when it is about science and will continue not to regardless of your advice.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
solo
International Hazard
Posts: 3975
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
Reference Information
Stereoselective and enantioselective synthesis of five-membered rings via conjugate additions of allylsulfone carbanions
Alfred Hassner †, Eugene Ghera, Tamar Yechezkel, Victoria Kleiman, Thiagarajan Balasubramanian
Pure Appl.Chem., Vol.72, No.9, pp.1671–1683, 2000
Abstract:
This lecture describes some of our studies of lithio derivatives of allyl sulfone carbanions which add α-regioselectively as well as anti
diastereoselectively to Michael acceptor olefins. This can be ascribed to chelation in the Michael addition step. When the reaction leads to
subsequent ring closure by using a bromoallyl sulfone, the latter acts as a methylenemethane synthon in a (3+2) Michael-initiated ring closure,
affording highly functionalized cyclopentane derivatives Such additions proceed with high stereoselectivity and with asymmetric induction leading to
nonracemic substituted cyclopentanones. Additions of allyl sulfone carbanions also proceed stereoselectively to C=N systems containing a chiral
auxiliary on N. These can be used in the synthesis of optically active five- and six-membered ring Nheterocycles. Furthermore, chiral groups on the
allyl sulfone moiety can induce significant remote asymmetric induction, made possible by the presence of an aromatic π-system which promotes
intramolecular chelation to the Li cation.
Attachment: Stereoselective and enantioselective synthesis of five-membered rings via conjugate additions of allylsulfone carbanions (130kB)
This file has been downloaded 1203 times
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
XxDaTxX
Hazard to Self
Posts: 66
Registered: 12-3-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Nicodem
PS: I never chill when it is about science and will continue not to regardless of your advice. |
It was a joke ... pun intended. The requisite temperature was -78oC, as in frozen HMPA.
[Edited on 10-12-2006 by XxDaTxX]
|
|
|
madcease
Harmless
Posts: 48
Registered: 9-9-2005
Member Is Offline
Mood: No Mood
|
|
Do you think it is possible to convert nitromethane into nitroethane?????
I have been doing some research into this and still unsure of how to go about.
|
|
|
SecretSquirrel
Hazard to Self
Posts: 71
Registered: 16-4-2006
Member Is Offline
Mood: No Mood
|
|
No, as far as I know nitromethane can't be converted into nitroethane by any reaction.
|
|
|
solo
International Hazard
Posts: 3975
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
| Quote: | Originally posted by Organikum
The direct reductionof phenylalanine to amphetamine is done with triethylsilane in trifluoroacetic acid IIRC. The silane being meanwhile a common
reagent AFAIK.
Reductions with this sytem almost always result in practically quantitative yields, thus beating the holy shit out of LiAlH. |
----------------------------------------------------------------------------------------
Selectivities in Ionic Reductions of Alcohols and Ketones with Triethyisilane / Trifluoroacetic Acid
Herbert Mayr and Barbara Dogan
Tetrahedron Letters, Vol. 38, No. 6, pp. 1013-1016, 1997
Abstract The relative rates of reduction of alcohols and ketones by Et3SiH/CF3CO2H have been de- termined by competition experiments
in order to derive scope and seleetivities of these reactions.
[Edited on 7-1-2007 by solo]
Attachment: Selectivities in Ionic Reductions of Alcohols and Ketones with Triethyisilane -Trifluoroacetic Acid .pdf (195kB)
This file has been downloaded 3622 times
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
Furch
Hazard to Others
Posts: 130
Registered: 8-10-2006
Member Is Offline
Mood: No Mood
|
|
I'm quite confident nitroethane can be made from nitromethane, using formaldehyde as methylating agent... Dehydrating the nitroalcohol with phtalic
anhydride and then reducing the formed nitroethene with a reduction agent of your choice.
I believe Organikum pointed out this way to go about the problem earlier in this thread... I.e. using formaldehyde to methylate phenylethylamine.
However the route is not only economically unfeasible, but also very inconvenient for such a simple molecule as amphetamine or nitroethane. Which is
to say, there are better and more interesting ways to make those substances...
... Which is why I came up with the idea of using MeI in the first place...
... Which later was "debunked" by most people in this thread :-P
Sincerely,
Furch
|
|
|
JohnWW
International Hazard
Posts: 2849
Registered: 27-7-2004
Location: New Zealand
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by solo
| Quote: | Originally posted by Organikum
The direct reductionof phenylalanine to amphetamine is done with triethylsilane in trifluoroacetic acid IIRC. The silane being meanwhile a common
reagent AFAIK.
Reductions with this sytem almost always result in practically quantitative yields, thus beating the holy shit out of LiAlH. |
Selectivities in Ionic Reductions of Alcohols and Ketones with Triethyisilane / Trifluoroacetic Acid
Herbert Mayr and Barbara Dogan
Tetrahedron Letters, Vol. 38, No. 6, pp. 1013-1016, 1997 |
Solo: Thanks for your effort, BUT that file cannot be downloaded in Windows because it has a colon in its file-name, wich is not allowed in Windows!
(You apparently use some other operating system). Please remove the colon from the name of the file, and re-upload.
|
|
|
tupence_hapeny
Hazard to Others
Posts: 131
Registered: 25-3-2007
Member Is Offline
Mood: continuing respiration (touch wood)
|
|
righto,
There appears to be some diehards on this topic, so I wish to do what I can...
Amino Acids can be reduced to amino aldehydes via the Akabori amino acid reduction with sodium amalgam:
http://www.chempensoftware.com/reactions/RXN006.htm
[ie. the second reaction: utilizes an ethanolic solution, saturated with HCl/Cl (I forget which) with sodium amalgam to reduce the amino acid to the
amino aldehyde (and esters of the same)].
I realise many here have no access to sodium metal or sodium amalgam, however, this synthesis from orgsyn uses electricity to generate the amalgam in
situ, in order to reduce the structurally similar cinnamic acid to hydrocinnamic acid:
http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv1...
The apparatus is as detailed in this preparation:
http://www.orgsyn.org/orgsyn/prep.asp?prep=CV1P0485
The reduction of the aldehyde to the alcohol is fairly straightforward (use whatever). The reduction of the alcohol to the alkane is achieved as per
the following:
http://www.erowid.org/archive/rhodium/chemistry/tyrosine2pma...
Which uses NaI (& thus HI) to generate the alkylhalide, reducing that with zinc.
Presumptions
Where I may be wrong
(1) As the electrolytic amalgam reduction will be carried out by virtually the route pioneered by Akabori et al, I don't think that it would cause the
loss of the amine group via hydrolysis.
(2) The amino aldehyde can be reduced by any known non-acidic (also non-strongly basic) route, such as Pt/C or Pd/C, etc.
(3) The amino alcohol will not undergo serious levels of hydrolysis by virtue of the NaI/HI (for the same reason ephedrine doesn't).
(4) The amine group could be removed and replaced prior to the reductive steps, maybe with methylamine, thereby avoiding P2P altogether (ie. H2SO4 to
remove the amine, oxidisation of the alcohol to the ketone - which will form an imine quicker than the carboxylic acid on the other arm of the
molecule). The reduction of the carbonyl group (Na/Hg) will also reduce the imine.
(5) The Na/Hg may also overreduce the aldehyde to the alcohol in one step, I am unsure of this, as I am not certain if the Na/Hg is capable of doing
so?
NB Attached is a brand new, MW reductive amination procedure - which uses Pt/C with in situ generated H2 (from aluminium powder) for the forming of
secondary amines from primary amines... Of some interest is the fact that Pt/C may be capable of doing all that is necessary in this procedure....
BTW, as to whether Nitromethane can be converted to Nitroethane, I found the following:
http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv5...
I have no idea how this would be made into nitroethane, however, it may in fact be possible (though how it would be done is beyond me - I see no way
to reduce the OH without reducing the NO2 - although it might be possible to work it via preferential shite)
[Edited on 24-4-2007 by tupence_hapeny]
Attachment: MW Reductive Amination (Pt on C) for synthesis of Secondary Amines (2006) 47 Tet Let 1437.pdf (82kB)
This file has been downloaded 1652 times
We are all the sum of our experiences, and our reactions to the same
|
|
|
formyl
Harmless
Posts: 1
Registered: 6-5-2007
Member Is Offline
Mood: No Mood
|
|
nitromethane can be alkylated via a dual deprotonation with 2 equiv n-butyllithium. once the second proton is removed, the most reactive site is at
carbon
|
|
|
| Pages:
1
2 |













