| Pages:
1
2 |
Magpie
lab constructor

Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
preparation of thymolphthalein
Introduction & Background
Thymolphthalein is an acid-base indicator that changes from clear to blue in the pH range 9.3 to 10.5. Use of this indicator is occasionally
specified in some of the old procedures such as those found in Organic Synthesis.
Thymolphthalein can be made via a Friedel-Crafts alkylation of phthalic anhydride with thymol:
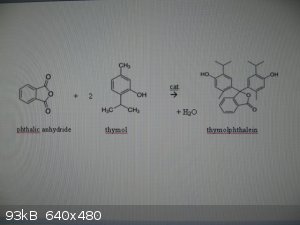
0. rx eqn
Thymol is the active ingredient in thyme, a common kitchen herb. To add some OTC to this synthesis I had originally planned to separate the thymol
from the oil extract of thyme, commonly available at health food stores. Then I read that ajowan oil was an Indian spice especially rich in thymol.
So I purchased 50ml planning to isolate the thymol by distillation. However, this was a failure as my ajowan oil did not contain any significant
amount of thymol based on the boiling points I obtained. I had purchased my ajowan oil from Dr Adorable. Perhaps this should have been my first
clue.
Next time around, not taking any chances, I purchased 25g of pure thymol. I have since found a supplier of thyme extract in Hungary claiming a very
high thymol percentage, but I did not buy this.
At first I could only find one very succinct and old procedure for making thymolphthalein (ref 1). It specified anhydrous ZnCl2 as the Lewis acid
catalyst. I tried this procedure using 10g of my thymol. It was a failure, resulting in a tarry mess.
I then decided to check my local university library where I can access all ACS journals. Here I found one article with a more detailed procedure (ref
2). Anhydrous SnCl4 was specified as the Lewis acid catalyst.
Experimental
A. Reaction
The procedure used was that provided in reference 2 at ½ scale. 15g of thymol (0.1 mole), 7.4g (0.05 mole) of phthalic anhydride, and 12.5g of SnCl4
were heated in an oil bath at 95°C while stirring. The SnCl4 was added over a period of 30 minutes. For this I used a 3-neck 100mL RBF as shown in
the picture below.

1. reaction set-up
The CaCl2 guard tube and the p-e funnel were not specified in the procedure but seemed appropriate to me. The temperature of the reaction was
monitored and controlled with a K type thermocouple and PID controller.
After the addition of the SnCl4 the flask was to be heated for an additional 30 minutes at 96°-103°C.
The temperature did get away from me rising to as high as 123°C.
B. Workup
The product was a very dark purple, hard, solid lump as shown below.

2. dark purple solid lump
To this was added to 100mL of 0.1M HCl and the stiff solid broken up with a spatula. It was somewhat difficult to break into pieces small enough to
be removed from the RBF.
The product was separated into the dark solids and a pink, flakey slurry. The dark pieces were ground to a sticky powder in a mortar. The pink
slurry of unknown identity was discarded; the dark powder was kept as the putative thymolphthalein. The picture below shows the two products.
[ 
3. rx products
After the mortar had been rinsed clean I placed it in my sink. I had made up a strong NaOH solution for testing. When I added this to the mortar it
turned an intensely dark blue color as shown below.

4. mortar with thymolphthalein residue and NaOH
The dark powder was washed several times with water using a 7cm Buchner funnel. This washing turned the powder a dull, light yellow as shown below.

5. powder washed with water
This powder was then dissolved in 350mL of acetic acid whereupon it turned an orange-red color as shown below.

6. thymolphthalein dissolved in acetic acid
The 350mL of thymolphthalein in acetic acid solution was then filtered, removing ~ 1/2g of white solids. The solids were thymophthalein (1st crop).
The filtrate was then distilled removing 225mL of acetic acid distillate per procedure. The 100mL of pot residue was now a deep carmine with no
solids present. This distillation is shown in the picture below.

7. Distillation of thymolphthalein dissolved in acetic acid
I attempted to get the thymolphthalein to precipitate by cooling the remaining 100mL of solution in an ice bath. But with the mp of acetic acid at
17°C this quickly froze. However, upon melting and setting overnight significant solid thymophthalein did precipitate (2nd crop). This was removed
by filtration. The filtrate is shown in the picture below.

8. 100mL of filtrate
The filtrate was then set up for a 2nd distillation. This time another 65mL of acetic acid was recovered. I expected to see significant
precipitation in the 35mL of pot residue after cooling but there was none. Again this compound showed it was capable of significant supersaturation.
Scratching the flask bottom with a glass stirring rod and adding thymolphthalein crystals did not cause precipitation. The flask was then left again
overnight. The next day revealed precipitated white solids on the bottom of the flask (3rd crop).
The 3 crops were allowed to dry at ~70°C. The 1st crop was ground to a fine powder in a mortar. The 2nd & 3rd crops were combined and ground to
a fine powder. Pictures of these products are shown below.

9. 3 crops of thymolphthalein precipitate

10. the ground product
Results
The mp of the 1st crop powder was 250°C. That for the combined 2nd & 3rd crops was 245°C. The wiki value is 248°- 252°C.
The yield was 0.50g of 1st crop, and 2.48g of the combined 2nd & 3rd crops, for a total of 2.98g. This gives a %yield of 13.8%.
A few mg of the product was dissolved in a ml of 95%ethanol. To this was added a few grains of solid NaOH. The solution turned an intense blue as
shown in the picture below.
Attachment: phpY53D7x (151kB)
This file has been downloaded 1061 times
11. thymophthalein in alcohol at high pH
Discussion
I have been working on this preparation for some time. My faux pas with the ajowan oil and poor first procedure (ref 1) were mostly
responsible for this.
The Hubacher procedure (ref 2) provides good detail for the reaction conditions. However, details for the workup were very limited. My choice of 3
distillations in retrospect seems excessive. But I really didn’t know what I was dealing with for sure as to how it was going to precipitate, etc,
so proceeded cautiously. This made for a tedious workup.
The yield was low but I did get the intended product. In picture 3 above the pinkish product in the beaker was discarded. It may have contained
significant product that I was not aware of. At any rate 3g will be a lifetime supply as it doesn’t take much to get a color change from clear to
blue.
It is interesting to note that Ex-Lax, once a popular laxative, was formerly made of phenolphthalein. Reference 2 is Hubacher’s evaluation of
carvacrolphthalein as a laxative. The declarative phrase “Smooth move Ex-Lax” can still be heard occasionally around my house when someone makes
a mistake.
References
1. “Practical Druggist and Pharmaceutical Review of Reviews,” Vol. 33 (1915).
2. “Carvacrolphthalein,” JACS, Vol. 64, Issue 11, pp. 2538-2539, [Contribution from the Laboratory of Ex-Lax, Inc], by Max H. Hubacher (Nov 1942)
http://pubs.acs.org/doi/abs/10.1021/ja01263a003?journalCode=...
Questions, comments, and suggestions are welcomed.
[Edited on 27-4-2016 by Magpie]
[Edited on 27-4-2016 by Magpie]
[Edited on 27-4-2016 by Magpie]
[Edited on 27-4-2016 by Magpie]
[Edited on 27-4-2016 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
Cool. I wish I had some. I would think it would be pretty easy to purify with chromatography.
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Nice work, Magpie!
It seems we share a quite wide field of interest regarding organic synthesis because what you do usually picks my interest too and/or something I
always wanted to do and maybe show to the public. :-) E.g. during the last few months I tried extracting carvacrol from a plant oil (but bought the
bad oil) to use in a synthesis instead of thymol. Then I came across a bottle of cymol (good precursor to carvacrol), but had no money with me to buy
it. :-) Maybe next time. :-)
Anyway, congratulations.
I felt a bit "premature" to discard that pinkish fluffy precipitate at the beginning of the workup and would surely postpone of the emptying of its
flask until I tried some sort of extraction on it. But you are right, 3 g pH indicator lasts a lifetime in an amateur laboratory.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
I remember reading somewhere that phenolphthalein could be prepared by condensing pthalic anhydride with phenol in the presence of conc
H2SO4 itself ? maybe the same could by done for thymol
anyways I found a paper claiming a 90% yield using methane sulfonic acid
http://www.sciencedirect.com/science/article/pii/S0040403909...
|
|
|
j_sum1
Administrator
Posts: 6335
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
Magpie. That's a great write-up and a nice little synthesis. I'm inspired.
I don't know how useful another indicator is, but it's a purty colour.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thank you JJay, Pumukli, and j_sum1.
Quote: Originally posted by CuReUS  | I remember reading somewhere that phenolphthalein could be prepared by condensing pthalic anhydride with phenol in the presence of conc
H2SO4 itself ? maybe the same could by done for thymol
|
Yes, I've seen this synthesis equation expressed with H2SO4 as catalyst, Wiki, IIRC.
I believe there is a synthesis of phenolphthalein in Brewster, also one for methyl orange indicator using phthalic anhydride.
I should mention that with this thymmolphthalein preparation as I was just adding the SnCl4 to the p-e funnel the thymol and phthalic anhydride were
being heated and the thymol had melted. At that time I saw the white/yellow reactants start to turn a very dark purple or black. At first I thought
"oh no! here comes the tar." But it was the first appearance of thymophthalein. I had not yet added any SnCl4. So, my point is: maybe the Lewis
acid is not all that necessary?
[Edited on 27-4-2016 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
CharlieA
National Hazard
Posts: 646
Registered: 11-8-2015
Location: Missouri, USA
Member Is Offline
Mood: No Mood
|
|
Great write-up, and great perseverance!
|
|
|
DraconicAcid
International Hazard
Posts: 4357
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Very nice.
Out of my field, but I would guess that similar reactions with phenol or cresol would give phenolphthalein and cresolphthalein (ETA: and
naptholphthalein]. I wonder how many other "..olphthaleins" one could make.
Hmmm...."The Carbon Compounds" (Porter, 1931) mentions this reaction on its chapter on dyes. Resorcinol reacts with phthalic anhydride to give
fluorescein, which can be brominated to give eosine. m-dimethylaminophenol will give rhodamine B.
[Edited on 27-4-2016 by DraconicAcid]
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
Nice writeup again. I like the photos and details. I wish JOC and J Med Chem required those... Very colorful work.
I can vouch that the preparation of phenolphthalein from phthalic anhydride with phenol in the presence of conc H2SO4 works, that was one of my first
experiments as a teenager. I did not test it as a laxative, however. I also made Bakelite, was was a giant mess, as it hardens into a glob that
was impossible to remove from anything it got on.
Fluoroscein is one of my favorite dyes to make and play with, a little goes a long way. I might still have some phthalic anhydride if anyone ever
needs some. Also, 3-nitrophthalic anhydride and hydrazine will make luminol, which is a great experiment to make and oxidize it to make light.
That and the dioxylates are some of my favorites chemoluminescent experiments as well.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Yes, I too have made a little fluorescein, which was fun because of its striking color. But making the resorcinol precursor was the real challenge.
Its precursor is the sodium salt of m-disulfonic acid which is made with a caustic fusion. Some time ago I had a metalworker make a copper crucible
especially for making this but I haven't used it yet. Someday I'll get back to it.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Superb work Magpie.
Very good write-up and inspiring photos.
Did you make the SnCl4 ?
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thank you aga. Yes, I made the SnCl4 a short time ago: http://www.sciencemadness.org/talk/viewthread.php?tid=28879
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
clearly_not_atara
International Hazard
Posts: 2800
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Why tin? At first I thought it was the easy synthesis, but you just used chlorine anyway.
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Nice work magpie, good to see someone still does real chemistry that is drug related.
A couple of questions though. In the original paper (reference 2 above) they recrystallised the crude material from acetic acid after leaching with
dilute HCl. The later presumably removes the tin salts. Why did you not follow this protocol? Why did you do an alkali extraction? I have found that
most phthaleins tend to oxidize in alkaline solutions so I would expect the alkali extraction to promote a certain amount of oxidation too.
My understanding of your procedure is that after the acid wash you decanted the flocculant precipitate and discarded it (mostly thymolphthalein?),
extracted the residue with alkali (in which thymolphthalein is pretty soluble and gives a solution that is prone to oxidation), is this correct? Did
you acidify the blue alkali extract to precipitate the thymolphthalein that went into solution?
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
I used SnCl4 since that was what the procedure called for. Toggle the link in reference 2 and you can read the Hubacher procedure.
Do you mean "not drug related"?
I never did an alkali extraction. I just washed it with water. The procedure said to wash it.
Edit: Maybe my use of the word "residue" in the mortar is confusing. What I meant was the traces of the dark purple gummy powder left in the mortar.
The ground up powder was taken up in acetic acid per procedure. I would clarify this in the OP but it is too late.
| Quote: Originally posted by Magpie |
After the mortar had been rinsed clean I placed it in my sink. I had made up a strong NaOH solution for testing. When I added this to the mortar it
turned an intensely dark blue color as shown below.
|
I do agree that I should probably have filtered/washed all of the products instead of just the dark purple gummy mass.
[Edited on 28-4-2016 by Magpie]
[Edited on 28-4-2016 by Magpie]
[Edited on 28-4-2016 by Magpie]
[Edited on 28-4-2016 by Magpie]
[Edited on 28-4-2016 by Magpie]
[Edited on 28-4-2016 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
Here is a GC(MS?) for genuine ajowan essential oil with peak IDs. As you can see, the real deal is about 40% thymol. In general, trust no "essential
oils" for sale on ebay or amazon. Fraud is rife and even reputable suppliers are sometimes taken for a ride (though by more sophisticated and harder
to detect methods). If it seems too cheap, there's a good reason for that (with few exceptions. some oils are just astoundingly cheap).
https://www.newdirectionsaromatics.com/msds/GCMS_AjowanEssen...
I am somewhat surprised that the ajowan you purchased was not authentic, however. It is an exceedingly cheap oil. The company that the GC is from
sells 100ml for $4.38 (They have a $100 order minimum, however).
Given the extremely low cost, perhaps ajowan essential oil is used to produce thymol that qualifies as a "natural flavor" (which commands a
significant premium over the synthetic material) and you were sold the forerun waste product. It looks like a fractionating the oil would leave a
stillpot residue of almost pure thymol after p-cymene passes over at 177C.
[Edited on 28-4-2016 by UC235]
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thanks for your research UC235.
Here's some excerpts from the notes I took during my distillation of Dr Adorable's ajowan oil:
"The ajowan oil from Dr Adorable is to be distilled by fractional distillation to attempt to obtain pure thymol. Other terpenes, etc, are expected to
be in the ajowan oil. 50% thymol would meet expectations. The bp of thymol is 232°C. A 100mL pot will be used. Ajowan oil charge of ~50mL of a
dark liquid.
Due to severe flooding shutdown and added magnetic stirring. First drop of condensate at 96°C. Quite a bit of condensate coming over at 170°C.
Shutdown - will do simple vacuum distillation tomorrow.
A lot of bumping - installed ebulliator. Went to packed column but too much flooding. Replaced packed column with Vigreux column in an effort to
eliminate flooding. Still too much flooding & foaming. Tried adding simethicone and DOT 5 silicone oil to eliminate foaming. Gave up due to
excessive foaming & flooding. There was only about 20mL in the pot. It didn't smell like thymol."
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
DraconicAcid
International Hazard
Posts: 4357
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by Magpie | Thanks for your research UC235.
Here's some excerpts from the notes I took during my distillation of Dr Adorable's ajowan oil:
"The ajowan oil from Dr Adorable is to be distilled by fractional distillation to attempt to obtain pure thymol. Other terpenes, etc, are expected to
be in the ajowan oil. 50% thymol would meet expectations. The bp of thymol is 232°C. A 100mL pot will be used. Ajowan oil charge of ~50mL of a
dark liquid.
|
Thymol is also a phenol, and terpenes are not. Why not extract with strong base?
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DraconicAcid | | Quote: Originally posted by Magpie | Thanks for your research UC235.
Here's some excerpts from the notes I took during my distillation of Dr Adorable's ajowan oil:
"The ajowan oil from Dr Adorable is to be distilled by fractional distillation to attempt to obtain pure thymol. Other terpenes, etc, are expected to
be in the ajowan oil. 50% thymol would meet expectations. The bp of thymol is 232°C. A 100mL pot will be used. Ajowan oil charge of ~50mL of a
dark liquid.
|
Thymol is also a phenol, and terpenes are not. Why not extract with strong base? |
Because that's too easy!
Incidentally, this is a good way to purify eugenol from clove oil as well.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
It seems that DraconicAcid is a better organic chemist than he purports. 
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
'Easy' sounds like a good plan to me ...
|
|
|
DraconicAcid
International Hazard
Posts: 4357
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Let's just say that I'm well-versed in first-year organic chem. Anything past that is a crap shoot.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Paddywhacker
Hazard to Others
Posts: 478
Registered: 28-2-2009
Member Is Offline
Mood: No Mood
|
|
Magpie, does the procedure for making Fluorescein not work for thymolphthalein: melting thymol and phthalic anhydride together with a drop of H2SO4
and holding the mixture molten for some time. I would have thought that the same procedure would work for phthalic anhydride and any phenol.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
I suspect you are right. Brewster (forum library) has a procedure. The big challenge is just making the resorcinol.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
gsd
National Hazard
Posts: 847
Registered: 18-8-2005
Member Is Offline
Mood: No Mood
|
|
As I understand it, Paddywhacker is suggesting that why not use the method used for making Fluoroscein to make Thymolphthalein. In making Fluoroscein,
98% H2SO4 (or Anhydrous ZnCl2) is used as dehydrating agent. Why not use C. H2SO4 as dehydrating agent for Thymolphthalein also?
gsd
On second thoughts I suspect H2SO4 will react with Thymol. Anhydrous ZnCl2 will not pose that problem it will act as dehydrating agent only.
BTW I have made Fluoroscein using both these reagents. They work like a charm.
gsd
|
|
|
| Pages:
1
2 |








