turd
National Hazard

Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
[Cooking] alkylthiohydroquinone dimethyl ether a.k.a. 1-alkylthio-2,5-dimethoxybenzene musings
Introductory remark 1: In my opinion there is too much "let's invite the law enforcement for coffee and cake"-style law-abiding chemistry going on. We
need more naughty chemistry and here's my attempt.
Introductory remark 2: Most of this is just recooking of known procedures with small adaptions. I know it's out of fashion, but repeating experiments
is supposed to be an integral part of science. Besides, if I can do it, everyone should be able to do it.
Introductory remark 3: The identity and adequate purity (for an amateur setting) of all products were asserted with physical methods. Due to personal
reasons I decided not to share this data.
1 Introduction
The 2,5-dimethoxy-4-alkylthio substitution pattern of phenethylamines has two representants in Shulgin's magical half-dozen [1], making it a highly
fascinating synthetic target. Shulgin himself used various ways to achieve this substitution pattern.
Aiming for 2C-T (the 4-methylthio compound), Shulgin went via sodium hydroquinonethiosulfate, the product of the addition of thiosulfate to
benzoquinone [2]. The thiosulfate was reduced with Zn/H<sup>+</sup> to mercaptohydroquinone (scheme 1). Mercaptohydroquinone was then
permethylated with dimethylsulfate. Besides the general nuisance of dissolving metal reductions, this scheme features one significant drawback making
it unsuitable for amateur settings, namely copious evolution of H<sub>2</sub>S. Indeed, Klute reported a serious poisoning by going this
route [3]. As an alternative Klute suggested acidic hydrolysis of the thiosulfate [4], but to my knowledge no success has been reported up to now.
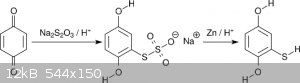
<i>Scheme 1</i>
For most non-methylthio compounds of the 2C-T family Shulgin alkylated 2,5-dimethoxythiophenol (actually the thiophenolate) with alkylhalides.
The dimethoxythiophenol was obtained by reacting hydroquinone dimethyl ether with chlorosulfuric acid followed by reduction with Zn (scheme 2) [5].
This scheme avoids formation of H<sub>2</sub>S but suffers instead from the use of chlorosulfuric acid, which is neither readily available
nor particularly agreeable to work with.
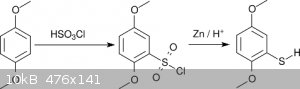
<i>Scheme 2</i>
To introduce S-substituents like <i>t</i>Bu or Ph, for which the corresponding halides are only poorly electrophilic, the alkyl
(aryl) disulfide was reacted with 2,5-dimethoxyphenyllithium (scheme 3) [6]. This procedure, albeit elegant, is difficult to perform in an amateur
setting, due to the need of dry reaction conditions. Moreover, producing the needed disulfides might be a stinky affair due to thioether side
products.
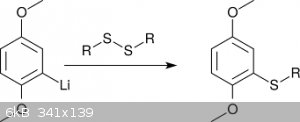
<i>Scheme 3</i>
In summary, the procedures performed by Shulgin are not very amateur friendly. Luckily, then came around Ullmann and reported a Duff formylation using
sulfuric instead of trifluoroacetic acid [7]. The thread was lauded for this OTC breakthrough, but it also contained fascinating sulfur chemistry. Two
points raised in this thread are of special interest:
Thiophenolates are significantly (by orders of magnitude) more nucleophilic than phenolates. Therefore, mercaptohydroquinone is also a useful
intermediate for alkylthiohydroquinone dimethyl ethers other than the S-Me analogue.
Mercaptohydroquinone can be made without dangerous or foul-smelling precursors or intermediates by addition of thiourea to benzoquinone giving
an isothiouronium salt. The salt is hydrolyzed first in acidic, then basic conditions (scheme 4). The intermediate is the bicyclic an virtually
odorless 5-hydroxybenzothioxolone (5-hydroxy-1-oxa-3-thia-2-indenone). This intermediate is especially interesting for target molecules with three
different alkyl substituents [7]. Notably, Shulgin was aware of this procedure and used it for the synthesis of 4T-MMDA-2 [8].
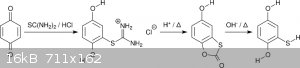
<i>Scheme 4</i>
Fascinated by this chemistry (and the pharmacology) I repeated these experiments in an attempt to make <i>n</i>-propylthiohydroquinone
dimethyl ether (1,4-dimethoxy-2-propylthiobenzene). Not all attempts were successful, but even failures can be instructive. Ultimately satisfying
yields were obtained.
In the thread of Ullmann there is suggestion of direct addition of thiols to benzoquinone. This is a more convergent synthesis, but it features the
drawback of having to go via a free thiol (available by alkylation of thiourea followed by hydrolysis). Moreover, the reaction may be more tricky than
it seems at a first glance because, depending on solvent, the thiol may add more than once [9]. Given the good yields of the linear synthesis
presented here, it's probably not worth it.
2 Results and Discussion
2.1 Mercaptohydroquinone
At first hydrolysis of hydroquinonethiosulfate was attempted. The best result was obtained by using perchloric acid in <i>n</i>PrOH, but
even there, besides mercaptohydroquinone, a large amount of red oil was obtained. Due to the potential formation of explosive alkylperchlorates [10]
this is highly discouraged.
Disappointed with the thiosulfate method, the thiourea method was tried instead. The first attempt can be considered a failure, though it is not 100%
clear where the error occurred. After addition of thiourea to benzoquinone in acidic conditions, a white precipitate of the isothiouronium salt is
supposed to appear, then conc. HCl is supposed to be added [7,8]. The precipitate did only appear after addition of the conc. HCl. Possible reasons
might have been a weighing error (the calculations were rechecked and are correct) or simply impatience (adding conc. HCl before cooling enough). The
method then calls for heating on a steam bath [7,8]. Being not familiar with this (archaic?) terminology, the reaction was heated at 70°C for 2 h. On
cooling a large mass of a precipitate was obtained, with a good nominal yield. Yet, surprisingly, analysis showed this to be single phase
isothiouronium chloride. The yield was low since this salt is mildly soluble in water and accordingly lots of material was lost by washing with water.
Since isothiouronium salts are classic intermediates in the synthesis of thiols there was no doubt that this can be hydrolyzed as well (scheme 5).
Indeed, refluxing for 2.5 h in aqueous NaOH seemed to afford marcaptohydroquinone in close to quantitative yields. Protection from atmosphere was
provided by a washing bottle with paraffin oil. No suckback was observed due to a constant evolution of NH<sub>3</sub> due to hydrolysis
of urea (scheme 5). Unfortunately, the mercaptohydroquinone was scorched when drying in vacuum at too high temperatures. A first recrystallization
attempt from MEK was a failure due to very high solubility. Toluene as recrystallization solvent was possible but also painful. On every step the
solution became significantly darker. Thus in total a mediocre yield was obtained (54% from benzoquinone, 84% for the basic hydrolysis).
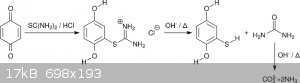
<i>Scheme 5</i>
On second attempt special care was taken to avoid errors of the first attempt. After the thiourea addition, the reaction was cooled to room
temperature and indeed a white precipitate of the isothiouronium salt was obtained. The reaction was then <i>refluxed</i> for one hour in
aq. HCl. After cooling, the benzothioxolone was filtered off, but not washed with water to avoid losses. It still contained some unreacted thiourea,
as was noted by evolution of ammonia during basic hydrolysis. Without recrystallization a satisfying yield of 83% (from benzoquinone) pure
mercaptohydroquinone was obtained.
The question thus arises, why go via the benzothioxolone for symmetric (with respect to both phenolic positions) substitution patterns? I'm quite sure
similar yields could be obtained without the cyclization step. It all depends on how quantitatively the isothiouronium salt can be made to
precipitates from the aqueous solution of first step (the addition reaction). On the other hand, the benzothioxolone is certainly a fascinating (and
nicely smelling) compound and gives you the bragging rights of having synthesized a heterocycle. So why not?
2.2 Alternative preparations of the benzothioxolone
Some people might not have access to thiourea. Fortunately there are alternative preparations. One interesting route starts with the
hydroquinonethiosulfate. It is reacted with a cyanide (a strong nucleophile) to thiocyanatohydroquinone. The latter is unstable and immediately
cyclizes to the imine derivative of 5-hydroxybenzothioxolone (scheme 6) [11]. This can be turned into the benzothioxolone by acid hydrolysis.
Certainly, it can also be hydrolyzed directly to mercaptohydroquinone in basic conditions. As a proof of concept I made the imine without further
hydrolysis. Since the hydroquinonethiosulfate had significantly decomposed on storage "only" 85 % were obtained. The original reference states near
quantitative yields [11], which I am sure are realistic
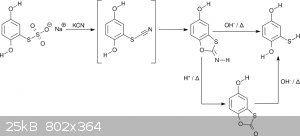
<i>Scheme 6</i>
Yet another, albeit low yielding, alternative is the reaction of hydroquinone or benzoquinone with inorganic thiocyanates [11].
2.3 Alkylation
The alkylation of the mercaptohydroquinone was performed in two steps: Propylation of the thiophenol followed by methylation of both phenols (Scheme
7).
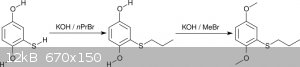
<i>Scheme 7</i>
The first step to <i>n</i>-propylthiohydroquinone proceeded in methanol with an excellent yield of 93% and high selectivity. No
purification was necessary to obtain a nicely crystalline and nearly colorless product. The high selectivity is based on a combination of two factors:
A - by orders of magnitude - higher acidity of the thiophenol functionality. Thus, by using approximately one equivalent of base there is little
phenoxide available at any time.
A - again by orders of magnitude - higher nucleophilicity of the thiophenol functionality.
These two points seem contradictory at first, after all nucleophiles are bases and the stronger acid forms the weaker conjugate base. But, whereas the
proton is a very hard acid, bromopropane is a soft electrophile. Therefore the proton preferentially protonates the hard phenoxide but the soft
thiophenoxide reacts quicker with bromopropane. Moreover, the thiophenoxide is probably distinctly less solvated in the alcoholic solvent, due to its
reduced ability of forming hydrogen bonds, leading to an even increased nucleophilicity.
Ullmann suggested the use of a weak base to completely avoid deprotonation of the phenoxides [7], but given the excellent selectivity of the protocol
presented here, this seems not to be necessary. Of course, it is crucial to use pure and dry reactants (mercaptohydroquinone and bromopropane) for
application of the correct stoichiometry.
The dimethylation of the <i>n</i>-propylthiohydroquinone was performed with MeBr using a method inspired by PainKilla [12]. Since Ullmann
reported problems with methylation of this substrate using MeI, the solvent was changed from MeOH to IPA, which increases the nucleophilicity of the
phenoxides as suggested by Nicodem [13]. Moreover, instead of bubbling the MeBr through the reaction mixture, it was condensed using a dry ice /
acetone bath and then added in one go to the reaction mixture. On one hand this allowed an estimation of the amount of MeBr used, on the other hand it
minimized oxidation with atmospheric oxygen. Yield was an acceptable 76%. There was a rather large amount of still acidic material. Sadly, no clean
mono-methylated product was isolated from this dark red oil.
The thus obtained <i>n</i>-propylthiohydroquinone dimethyl ether was a tan crystalline material with probably adequate purity for
subsequent reactions. Nevertheless, since the melt was very dark, it was considered appropriate to distill it. Distillation at <1 mbar proceeded
without problems and 95% of a colorless oil, which spontaneously crystallized to a glistening white mass, was recovered.
3 Conclusion
The sulfur chemistry brought to our attention by Ullmann is most fascinating. And it is clearly not restricted to a professional setting, so go ahead
and cook. If the proper precautions are taken, there has to be no fear of (literally) nosy neighbours. Perhaps most importantly, this work is further
proof of the futility of prohibition. Don't let nobody stop you.
4 Experimental
4.1 General
No special glassware (like multi-neck flasks) was used. Inert atmosphere was provided by bubbling Ar and quick stoppering. Chemicals were drugstore
(technical) quality. Benzoquinone was made like detailed in a thread here [14] and dried over night in vacuum. <i>n</i>-Propyl bromide was
made from HBr and n-propanol. It was dried over MgSO<sub>4</sub> and fractionated before use.
4.2 Mercaptohydroquinone, failed run
93 g (860 mmol, 1 eq.) benzoquinone was dissolved in 500 ml AcOH and slowly dropped into a solution of 73 g (0.950 mmol, 1.1 eq) thiourea in 700 ml
2.5 N HCl (146 ml conc. HCl diluted to 700 ml, 1750 mmol). After stirring for a few minutes 44 ml conc. HCl (528 mmol) were added to the clear
solution, resulting in a white precipitate. The reaction was heated at 70°C for 2 h, whereby the precipitate redissolved to a clear orange solution.
The reaction was cooled over night at 4°C, the precipitate filtered off, washed with cold H<sub>2</sub>O and dried over night in vacuum.
Yield: 121.5 g (64 %) isothiouronium chloride.
A round bottom flask with a stir bar was charged with 110 g NaOH (2750 mmol, 5 eq.) and 1 l water. After dissolution of the NaOH, vacuum (ca. 20 mbar)
was applied for 5 min. Ar was then bubbled through the solution for ca. 5 min. 121.5 g (551 mmol, 1 eq.) isothiouronium chloride was added while still
bubbling Ar. The flask was quickly connected to a reflux condenser, which was in turn connected to a washing bottle with paraffin oil as protection
from the atmosphere. The reaction was stirred and refluxed, whereby it turned from yellow to translucent green. There was a steady evolution of gas
(ammonia), thus no problems with suckback. After 2.5 h the condenser was removed and the flask immediately stoppered to avoid contact with air. After
cooling to room temperature stirring was continued and slowly conc. HCl was added until acidic to universal indicator paper. Ca. 240 ml conc. HCl were
needed. The reaction turns from green to red to translucent yellow and there is notable fuming due to formation of NH4Cl. The reaction was extracted
with EtOAc (DCM is not effective, but gives a very clean product, thus continuous extraction with DCM might be a good idea for those with the
appropriate glassware), the organic phase washed with water and dried over Na<sub>2</sub>SO<sub>4</sub> and evaporated to
dryness, giving a tan powder. Unfortunately there was scorching while attempting to dry in vacuum and thus after a few miserable recrystallization
attempts 66 g (84% from the isothiouronium chloride or 54 % from benzoquinone) were obtained.
4.3 Mercaptohydroquinone, successful run
93 g (860 mmol, 1 eq) benzoquinone was dissolved in 500 ml AcOH and slowly dropped into a solution of 73 g (0.950 mmol, 1.1 eq) thiourea in 700 ml 2.5
N HCl (150 ml conc. HCl diluted to 700 ml, 1800 mmol). Stirring was continued until the reaction was cooled to room temperature. A large amount of
white precipitate formed. 44 ml conc. HCl (528 mmol) was added and the reaction was refluxed for 1 h, whereby the precipitate redissolved. The
reaction was cooled over night at 4°C and the precipitate filtered off and sucked dry to give translucent colourless needles.
A freshly prepared solution of 137 g NaOH (3425 mmol, 4 eq.) in 1 l H<sub>2</sub>O was kept under vacuum (ca. 20 mbar) for 5 min and then
bubbled for 5 min with Ar. The whole precipitate from the last step was added (while maintaining bubbling of Ar) and the flask quickly attached to a
reflux condenser, which was in turn connected to a washing bottle with paraffin oil. The reaction was refluxed for 1 h, whereby it turned from yellow
to greenish yellow. There was minor evolution of NH<sub>3</sub> due to decomposition of unreacted thiourea adsorbed to the precipitate.
The condenser was then removed and the flask quickly stoppered and cooled in a stream of cold water (bubbling due to evaporation of
NH<sub>3</sub> . 370 ml conc. HCl was then added while stirring (color
changes from greenish yellow to red to pale yellow). The reaction was filtered to remove a few black specks and extracted thrice with EtOAc.
Attention: On first extraction the organic phase was heavier than the aqueous phase, leading to an interesting lava lamp effect. The organic phase
would first rise, slowly saturate with product and then sink again. The organic phase was washed with water and brine and dried over
Na<sub>2</sub>SO<sub>4</sub>. The volatiles were removed under vacuum and the residue dried over night at 40°C to give 101.4
g (83 % from benzoquinone) of a solid ranging from shiny white needles (first precipitate) to pale yellow powder (last precipitate). . 370 ml conc. HCl was then added while stirring (color
changes from greenish yellow to red to pale yellow). The reaction was filtered to remove a few black specks and extracted thrice with EtOAc.
Attention: On first extraction the organic phase was heavier than the aqueous phase, leading to an interesting lava lamp effect. The organic phase
would first rise, slowly saturate with product and then sink again. The organic phase was washed with water and brine and dried over
Na<sub>2</sub>SO<sub>4</sub>. The volatiles were removed under vacuum and the residue dried over night at 40°C to give 101.4
g (83 % from benzoquinone) of a solid ranging from shiny white needles (first precipitate) to pale yellow powder (last precipitate).
4.4 n-Propylthiohydroquinone
27.1 g (411 mmol, 1.05 eq.) 85% KOH was dissolved in 300 ml MeOH and degassed by application of vacuum (ca. 20 mbar, 5 min) and bubbling with Ar (5
min). 55.6 g (392 mmol, 1 eq.) mercaptohydroquinone were then added and the flask quickly stoppered. The reaction was stirred until full dissolution
(bright yellow solution). 50.6 (411 mmol, 1.05 eq) <i>n</i>PrBr were then quickly added under stirring and the flask stoppered tightly.
There was immediate deposition of white KBr and a distinctly exothermic reaction. Fearing lower selectivity due to increased temperature, the reaction
was cooled in a water bath and stirring continued over night. The reaction was acidified with a small amount of conc. HCl and the inorganics removed
by filtration. The filter cake was washed with a small amount of MeOH. The volatiles of the combined organic phases were removed in vacuum and the
residue partitioned between EtOAc and H<sub>2</sub>O, which proved difficult due to a nearly identical density of both phases. The aqueous
phase was extracted with a small amount of EtOAc and the combined organics were washed with a small amount of brine, dried over
Na<sub>2</sub>SO<sub>4</sub> and evaporated to dryness in vacuum. The residue was dried over night in vacuum to give 67.3 g
(93 %) of tan solids.
4.5 n-Propylthiohydroquinone dimethyl ether (2,5-dimethoxy-1-propylthiobenzene)
40 g 85% KOH (606 mmol, 2.2 eq.) was dissolved in 400 ml IPA by stirring over night in a stoppered flask. The solution was degassed by application of
vacuum (ca. 20 mbar, 5 min) and bubbling with Ar (5 min).
50 g (270 mmol, 1 eq.) <i>n</i>-propylthiohydroquinone was added and the flask quickly stoppered. After stirring for a few min, a greenish
yellow, slightly turbid solution was obtained. The greenish tint is possibly caused by oxidation products - even the slightest contact with air
produces dark green smears. This solution can be stored for days without notable decomposition.
A round bottom flask was marked at 60 ml, corresponding to ca. 102 g (1074 mmol, 4 eq.) of MeBr. It was stoppered with a two-holed rubber stopper. One
hole was fitted with a glass tube extending down to the bottom, the second hole by a glas tube only extending to slightly below the stopper. The long
glass tube was connected with rubber tubing to the vacuum adapter of a distillation setup. The short glass tube was connected to a washing bottle with
paraffin oil. The distillation flask was filled with a suspension of 160 g (1344 mmol, 5 eq.) dried KBr in 140 ml H<sub>2</sub>O. The
suspension was degassed by application of vacuum (ca. 20 mbar, 5 min) and bubbling with Ar (5 min). The receiving flask was empty and cold water was
run through the condenser. The thermometer was replaced by a dropping funnel containing a solution of 148 g (1510 mmol 5.6 eq.) of 95%
H<sub>2</sub>SO<sub>4</sub> in 62 g (1938 mmol, 7.2 eq.) MeOH, degassed by bubbling Ar for 5 min. The distillation flask was
heated to a gentle boil and the MeOH/H<sub>2</sub>SO<sub>4</sub> solution slowly dropped in. After a minute of gas generation
(bubbling in the washing bottle), the round bottom flask with the two-holed rubber stopper was immersed into an acetone/dry ice bath. After all
MeOH/H<sub>2</sub>SO<sub>4</sub> had been added, heating was continued until the 60 ml mark was reached (ca. 2 h - the
reaction is surprisingly slow). There is little to no bubbling in the washing bottle during the reaction since all MeBr is condensed.
The rubber stopper was then replaced by a ground glass stopper (while still immersed in the acetone/dry ice bath). The MeBr was quickly added to the
well stirred IPA solution of the dipotassium salt of <i>n</i>-propylthiohydroquinone and the flask immediately stoppered and clamped
tight. There was no notable problem with overpressure. The reaction turned immediately into a green suspension closely resembling a matcha latte.
After ca. 30 min a yellow suspension was obtained. Large masses of clumpy KBr precipitate make a good stirbar essential. The reaction was stirred over
night (stirring becomes easier due to finer KBr), the inorganics filtered off, washed with IPA and the filtrates combined.
Around two third of the IPA were removed in vacuum and the solution diluted with H<sub>2</sub>O and extracted thrice with DCM. The
combined organics were washed with ca. 10 % NaOH (aqueous phase at the bottom!) and brine, dried over
Na<sub>2</sub>SO<sub>4</sub> and the volatiles removed in vacuum. A pale yellow solid (44 g, 76%) was obtained.
The combined aqueous phases were acidified with conc. HCl (pH~1), extracted with DCM, dried over Na<sub>2</sub>SO<sub>4</sub>
and the volatiles removed in vacuum. A dark red oil was thus obtained which did not crystallize and eventually was dumped.
Since the melt and solutions of the <i>n</i>-propylthiohydroquinone dimethyl ether were quite dark (three consecutive reactions without
purification by crystallization or distillation), the combined products of two runs were distilled (115-130°C, < 1 mbar) with a recovery of 95% to
give a virtually colorless oil that spontaneously crystallized to shiny white needles.
4.6 5-Hydroxybenzothioxolone imine (2-imino-1-oxa-3-thia-5-indenol)
Sodium hydroquinonethiosulfate was prepared as described by Shulgin [2] and isolated as the monohydrate. After standing for ca. 6 months it had
darkened considerably. 67.5 g (256 mmol, 1 eq.) of such impure sodium hydroquinonethiosulfate hydrate was stirred for 30 min in 400 ml
H<sub>2</sub>O and filtered to give a dark brown solution. 23 g (361 mmol, 1.4 eq) KCN dissolved in a small amount of
H<sub>2</sub>O was added while stirring. A tan precipitate deposited immediately and stirring was continued for 2 h. The precipitate was
filtered off, washed with water until washings were colorless and dried overnight in vacuum at ca. 40°C. 36.5 g (85%) of the imine was obtained as a
tan powder.
Acknowledgements
This work is dedicated to all active amateur chemists and would not have been possible without the inspirational work of (neither in particular order
nor comprehensive) Klute, Ullmann and PainKilla. A very special thank you goes out to Nicodem for the gargantuan patience with us ignoramuses and for
the Sisyphean task of spreading knowledge.
References
[1] A. Shulgin and A. Shulgin, PiHKAL
[2] A. Shulgin, PiHKAL#39
[3] Klute, https://www.sciencemadness.org/whisper/viewthread.php?tid=10...
[4] Klute, https://www.sciencemadness.org/whisper/viewthread.php?tid=11...
[5] A. Shulgin, PiHKAL#40
[6] A. Shulgin, PiHKAL#6
[7] Ullmann, https://www.sciencemadness.org/whisper/viewthread.php?tid=11...
[8] A. Shulgin, PiHKAL#167
[9] A. R. Katritzky, D. Fedoseyenko, O. O. Mohapatra and P. J. Steel, Synthesis 2008, 777–787 (2008)
[10] https://www.sciencemadness.org/whisper/viewthread.php?tid=10...
[11] H. Fiedler, Chem. Ber. 95, 1771–1785 (1962)
[12] PainKilla, https://www.sciencemadness.org/whisper/viewthread.php?tid=10...
[13] Nicodem, https://www.sciencemadness.org/whisper/viewthread.php?tid=10...
[14] https://www.sciencemadness.org/whisper/viewthread.php?tid=14...
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
first of all,I must thank you,turd,for starting this thread.As I love syntheses a lot,this thread is a sight for my sore eyes.Now,a few questions,if
you don't mind
| Quote: | | Two points raised in this thread are of special interest:...Thiophenolates are significantly (by orders of magnitude) more nucleophilic than
phenolates |
isn't that obvious ,going by the periodic trends of group 16.
| Quote: | Indeed, refluxing for 2.5 h in aqueous NaOH seemed to afford marcaptohydroquinone in close to quantitative yields...
84% for the basic hydrolysis |
don't think of me as being rude,but I am sure other members would agree that 84% is not "close" to quantitative yields.But the hydrolysis could be
made more efficient and the reaction time reduced with certain modifications
| Quote: | | Some people might not have access to thiourea |
thiourea can be made,I have seen an prep on organic syntheses.Sadly I can't find it now.
recently I read a paper in which aryl halides were converted to phenols in high yields using copper halide catalyst,KOH and PEG-400(polyethylene
glycol is sold as laxative).If KOH is replaced by KSH,maybe thiophenols could be obtained.
benzoquinone can be reacted with HBr to get the bromo hydroquinone,or hydroquinone methyl ether could be brominated using NBS,and then the halide can
be replaced with SH.
http://www.sciencedirect.com/science/article/pii/S1566736711...
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
Thank you for your comments. Any constructive criticism is very welcome.
Fair enough, but interesting and obvious aren't mutually exclusive...
Quote: Originally posted by CuReUS  | | Quote: | Indeed, refluxing for 2.5 h in aqueous NaOH seemed to afford marcaptohydroquinone in close to quantitative yields...
84% for the basic hydrolysis |
don't think of me as being rude,but I am sure other members would agree that 84% is not "close" to quantitative yields.But the hydrolysis could be
made more efficient and the reaction time reduced with certain modifications
|
Indeed, 84% is not close to quantitative, that's why I wrote "seemed". The rather low isolated yield was
due to scorching and problems with recrystallization. The still wet product was not dark at all and the acidified solution pale yellow. It seems that
mercaptohydroquinone takes the hydrolysis conditions (refluxing NaOH under inert atmosphere) surprisingly well. That of course raises the question how
comes that it got significantly darker on recrystallization? That might be a problem of my hardware store solvents. Indeed, my toluene turns deep red
when refluxed with tosic acid on a Dean-Stark trap.
| Quote: Originally posted by CuReUS | | recently I read a paper in which aryl halides were converted to phenols in high yields using copper halide catalyst,KOH and PEG-400(polyethylene
glycol is sold as laxative).If KOH is replaced by KSH,maybe thiophenols could be obtained. |
If you go via a halide, you could directly add an alcohol or perhaps even a thiol. See for example https://www.sciencemadness.org/whisper/viewthread.php?tid=70.... I was never 100% happy with this method, because I always got some unreacted
bromide, which was a pita to remove.
BTW, I have a bottle of NaHS, but never open it, because.... Ewwwww....
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Cureus, I'm sure you are talking about alkylated thiourea http://www.orgsyn.org/demo.aspx?prep=CV3P0617 which is a well know rearrangement (thiocyanate <-> thiourea).
NaSH/KSH is a relatively weak base. This is why it is a good acceptor for alkyls. C-S is a coavalent bound, while C-O in phenolates and alkoxide is
polar.
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
I must be ignorant because I don't know what makes this product/methodology edgy or whatever. Either way this is a really well written post and
features a nice compendium of other's and you're own work. Excellent job  . You
could have easily prepubbed this, but people would probably yell for analysis. I trust you did your due dilligence given the quality of the report. . You
could have easily prepubbed this, but people would probably yell for analysis. I trust you did your due dilligence given the quality of the report.
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
Thanks. Concerning purity it has to be noted that without proper Schlenk techniques a small amount of oxidation is unavoidable, as is some oligomeric
product. Thus, with the exception of the final distilled product, nowadays you could not publish this in a high-impact synthesis journal [except by
application of the usual tickery as purposefully low resolution spectra, spectra and yield from different runs, etc.].
Nevertheless, I was pleased by the quality of the products and the impurities seem to be benign as shown by the final distillation with 95% recovery.
I think the overall yield for this four step (five, if counting the cyclization step) procedure is quite respectable.
Perhaps a small note for the olfactory connoiseurs: The propylthioydroquinone has a nice fresh and fruity smell. The dimethyl ether still smells
fruity but has a distinct garlic component. Although I like both, the combination is a bit uncanny.
|
|
|
Crowfjord
Hazard to Others
Posts: 390
Registered: 20-1-2013
Location: Pacific Northwest
Member Is Offline
Mood: Ever so slowly crystallizing...
|
|
Good work, turd. You mentioned having tried hydrolysis of the hydroquinonethiosulfate, with perchloric acid giving the best results. Which other
acids did you try? Did you slowly add the arylthiosulfate to the hot acid, or do an all-at-once sort of thing? I read in Klute's thread that a competing reaction with hydrolysis is reaction of the organic thiosulfate with the thiol, which forms a disulfide. Perhaps
this was a constituent of your side products. I am curious, as I had previously looked and failed to find much info on such a reaction; I was going to
do some experiments on the matter myself, but since you have now given it a shot, now I can pick your brain
Also, thanks for the organoleptic data, I am personally a big fan of laboratory smells and stinks.
[Edited on 25-6-2015 by Crowfjord]
[Edited on 25-6-2015 by Crowfjord]
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
Sorry, I fear I'm not going to be much help. These were not systematic experiments, but random shots. I tried aq. HCl, aq.
HClO<sub>4</sub>,HClO<sub>4</sub> in propanol (not recommended!), HClO<sub>4</sub> in THF (not recommended!),
H<sub>2</sub>SO<sub>4</sub> in propanol. Typically at 50°C ranging from 2 h to over night.
In all cases complex mixtures were obtained. Isolated compounds were hydroquinone, S<sub>8</sub>, mercaptohydroquinone, unreacted Bunte
salt, S-alkylated products (!) and always unidentified oil.
There is some literature on the hydrolysis of Bunte salts, mostly kinetic studies, e.g. JACS 88, 5245-5250 (1966).
If you get this to work, this would be fantastic, but expect some pain. If you have access to cyanides I would recommend to just react the thiosulfate
with that. It's a very clean reaction.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
you have hit the nail on the head.Thank you
turd,I have a few questions more
| Quote: | | In the thread of Ullmann there is suggestion of direct addition of thiols to benzoquinone. This is a more convergent synthesis, but it features the
drawback of having to go via a free thiol (available by alkylation of thiourea followed by hydrolysis). |
I was thinking,why not do the hydrolysis and the alkylation in one pot.That way,you wouldn't have to handle the free thiol.
also,can't you make thoiethers from sulphide ions.Mix Benzoquinone and Na2S and drip in n-propylbromide.
| Quote: | | There is some literature on the hydrolysis of Bunte salts...If you get this to work, this would be fantastic |
dammit,I was going to tell you about that.there is a thread on the synthesis of modafinil on SM where bunte salt was used
http://www.sciencemadness.org/whisper/viewthread.php?tid=611...
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Sad to say, but https://www.sciencemadness.org/whisper/viewthread.php?tid=70... is most likely a bullshit (aryl-halide to phenol via PEG+CuI):
CuI + NaOH => CuOH + NaI
Usually for ullmann reaction you take something like CuI+K2CO3. And on boling Cu(I) turns into Cu+Cu(II):
2CuI => Cu + CuI2
|
|
|
Draco_440
Harmless
Posts: 2
Registered: 22-9-2006
Member Is Offline
Mood: No Mood
|
|
Why not form 2,5-Benzaldehyde then go this route? https://www.erowid.org/archive/rhodium/chemistry/4-alkylthio...
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Late update: I was talking about this article in my previuos message http://www.sciencedirect.com/science/article/pii/S1566736711... , modafinil preparation has nothing to do with it.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Draco_440, deactivated 1,4-benzoquinones give only 3-substituted products:
http://www.sciencedirect.com/science/article/pii/S0040402001...
http://www.sciencedirect.com/science/article/pii/S0040402001...
http://www.sciencedirect.com/science/article/pii/S0968089603...
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
byko,calm down.You are saying that its bullshit,but have you tried it out ? .I don't think scientists are mad to publish something in a reputed
journal just for fun.Also if you search the autthors,you will find that they have been playing around with CuI for a long time.This is not their first
paper.
| Quote: | | modafinil preparation has nothing to do with it. |
what do you mean ? the CuI paper has obviously nothing to do with modafil,but the bunte salt method could be using to make aryl thio compounds as
well.
|
|
|
Scr0t
Hazard to Others
Posts: 118
Registered: 14-1-2012
Location: Europe
Member Is Offline
Mood: Desiccated
|
|
I see in your successful run in reacting benzoquinone with thiourea you added additional aq. HCl and heated for a further amount of time.
When I did this as described by Shulgin under 4T-MMDA-2, it was heated at 93-96°C. The benzothioxolone was obtained in excellent yields of 94% as
large off-white needles. Subsequent runs were similar. IIRC the yield was better than Shulgin's reported yield.
The only difference in the quantities used was the benzoquinone needed a bit more AcOH solvent to dissolve due to somewhat low ambient temperatures.
Methylation of the hydroquinone though was done in DMF with MeI (18hrs @75°C) and only got yields in the ballpark of 60% of the dimethyl-ether for
various alkyl groups on the sulphur atom.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Turd, thanks for the excellent report. I don't have much to say about your work other than it was a pleasure to read about it.
I'm attaching the review article Angew. Chem. Int. Ed. 1967, 6, 544–553, DOI: 10.1002/anie.196705441 (Klute cited it in his thread
and I think it is worth making it more available). I also think the reductive cleavage of Bunte salts is not yet as well researched as it could be.
There are chances that it could be performed under basic media with inexpensive and available sulfur-based reducing reagents
(Na2S2O4, thiourea dioxide, Na2S, etc.). Transthiolation with sacrificial mercaptanes (e.g.,
thioglicolic acid) could also be another possibility. Direct nucleophilic substitution with NaBH4 in a proper solvent could also do the
job. In principle, there is potential for amateurs developing new methods for this reaction.
On a Bunte salts related context it might be worth mentioning that 2,5-dimethoxy-1-propylthiobenzene could potentially be made by coupling a propyl
Bunte salt with 2,5-dimethoxyphenylmagnesium halide (DOI: 10.1021/ol500067f). Though in my opinion, directed ortho-lithiation of
1,4-dimethoxybenzene followed by the reaction with elemental sulfur might be a much more practical route to 2,5-dimethoxyphenyl alkyl thioethers,
particularly if it turns out that the so formed lithium thiophenolate could be S-alkylated in a one-pot fashion, thus avoiding the isolation
of the thiophenol intermediate.
Attachment: The chemistry of Bunte salts.pdf (365kB)
This file has been downloaded 2569 times
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
|








