ParadoxChem126
Hazard to Others

Posts: 104
Registered: 5-4-2013
Location: USA
Member Is Offline
Mood: No Mood
|
|
Chlorobenzene from Aniline via the Sandmeyer Reaction
August 21, 2014
By ParadoxChem126
Introduction
Chlorobenzene is a useful chemical in organic syntheses. It dissolves a wide range of organic compounds, making it an effective high-boiling solvent.
It also has numerous applications as a reagent. For example, it is a direct precursor to phenylmagnesium chloride, a valuable Grignard reagent that
can be used to produce a variety of compounds (e.g. benzene, triphenylmethanol, etc.).
The most common laboratory preparation of chlorobenzene involves the direct chlorination of benzene using a Lewis acid catalyst. This preparation is
considerably hazardous, as it involves the use of very toxic and carcinogenic chemicals. For home chemists, a safer method of preparing chlorobenzene
is the use of the Sandmeyer reaction. This reaction entails the diazotization of aniline to a phenyldiazonium salt, followed by the catalytic
decomposition of that salt with copper(I) chloride. The main resultant product is chlorobenzene.
In this experiment, chlorobenzene will be prepared from aniline via the Sandmeyer reaction, as detailed above. This is a slightly modified,
scaled-down version of the preparation found on page 577 of Vogel’s Textbook of Practical Organic Chemistry, Second Edition.
Experimental
Combine 20 mL of aniline and 57 mL of distilled water in a 600 mL beaker. Place the beaker on a magnetic stirrer, and slowly add 57 mL of 37%
hydrochloric acid to the beaker in small portions. Small amounts of white fumes are evolved.
Addition of hydrochloric acid to the reaction mixture
Once the fumes subside, place the beaker in a salt-ice bath and wait for the temperature to drop to 0°C or below. A white precipitate of aniline
hydrochloride is observed at this low temperature.
Once the mixture reaches the desired temperature, begin to add a solution of 16g of sodium nitrite in 33 mL of distilled water using an addition
funnel. This forms nitrous acid in situ. Add the sodium nitrite solution dropwise to ensure that the temperature stays as low as possible.
Do not let the temperature rise above 5°C, as the sensitive phenyldiazonium chloride would decompose. If the temperature gets too high, brown-orange
nitrogen dioxide gas will be released. Keep the magnetic stirrer stirring and use the salt-ice bath for cooling throughout the entire addition. The
white precipitate of aniline hydrochloride dissolves as it reacts with the nitrous acid.
Addition of sodium nitrite solution to the reaction mixture
Meanwhile, dissolve 27.72g of copper(I) chloride in 113 mL of 37% hydrochloric acid in a 1 L three neck round-bottom flask. The resulting solution is
dark green to black. Place the flask in a salt-ice bath on top of a magnetic stirrer. Allow the temperature to drop to 0°C or below.
When the sodium nitrite addition is complete, allow the mixture to finish reacting for a few minutes. Use the salt-ice bath to keep the temperature
as low as possible. Then, begin to slowly add the phenyldiazonium chloride solution to the copper(I) chloride solution in small portions. Keep the
phenyldiazonium chloride cold between additions.
Addition of the phenyldiazonium chloride to the copper(I) chloride
The mixture thickens due to the separation of an addition product between the diazonium salt and the copper(I) chloride. During the addition of the
diazonium salt, small amounts of brown-orange nitrogen dioxide gas are released. Once all of the diazonium salt has been added, remove the cooling
bath and allow the mixture to come to room temperature without external heating. The mixture becomes less viscous as it warms up. During this step,
nitrogen dioxide gas is released, so it is necessary to do this outside or under a fume hood.
Evolution of nitrogen dioxide gas
As the mixture warms up, effervescence is observed as nitrogen gas (N2) is released. By the time the reaction reaches room temperature,
the evolution of brown-orange nitrogen dioxide will have stopped. Once the mixture reaches room temperature, a hot water bath is applied to slowly
bring the reaction up to 60°C. Magnetic stirring is continued throughout the heating process. The release of nitrogen gas continues for several
minutes.
Heating the reaction mixture on a hot water bath
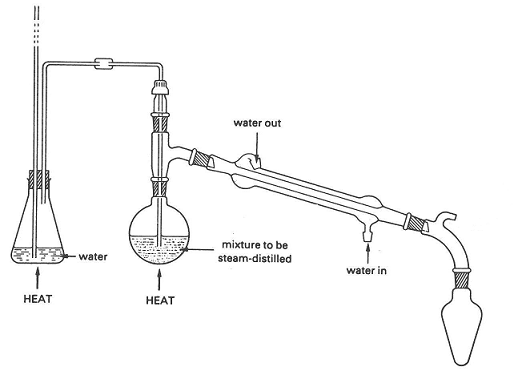
Once no more nitrogen is evolved, set up for steam distillation using the following (or similar) apparatus:
Apparatus for steam distillation
The leftmost flask contains distilled water that is boiled to generate steam. The steam travels up the tube and bubbles through the reaction mixture.
The resulting steam/vapor mixture is condensed in the condenser and the distillate is stored in the rightmost flask. It is important to note that
the reaction flask must be heated; otherwise, the steam will simply condense upon contact with the cooler reaction mixture.
As the distillation commences, drops of yellow oil will appear in the distillate. This oil is the crude chlorobenzene. Continue the distillation
until no more oily drops are present in the distillate.
Steam distillation to separate the crude chlorobenzene
The distillate was given time to form two layers. Then, it was transferred to a separatory funnel and the layers were separated. Discard the upper
aqueous layer and keep the lower chlorobenzene layer. Transfer the chlorobenzene back to the separatory funnel and wash it twice consecutively with
solutions of 2.5g of sodium hydroxide in 25 mL of water. The sodium hydroxide solution helps to remove phenol and other side products. Wash the
chlorobenzene one more time with 30 mL of distilled water.
Chlorobenzene (bottom) and water (top) after distilled water wash
Transfer the chlorobenzene to a 50 mL Erlenmeyer flask and add enough anhydrous calcium chloride until the CaCl2 is free-flowing without
clumps. This removes any traces of water from the chlorobenzene. Place the flask on a magnetic stirrer and stir it for about 30 minutes to complete
the drying process.
Decant the chlorobenzene into a 100 mL round-bottom flask. Rinse the Erlenmeyer flask and calcium chloride with a total of 35 mL of dry
dichloromethane in small portions. Add all of the dichloromethane rinses to the 100 mL round-bottom flask. Rinsing with dichloromethane ensures that
none of the chlorobenzene is lost on the desiccant.
Distillation of the dichloromethane/chlorobenzene mixture
Set up for simple distillation and distill off all but about 1 mL of liquid. The distillate is a colorless mixture of dichloromethane and
chlorobenzene. The distillate, contained in a 125 mL round-bottom flask, is placed on a hotplate set to very low heat. The goal is not to boil the
solution; it is to only provide enough heat to aid in evaporation. Once the schlieren due to dichloromethane vapor are no longer visible, continue to
heat for another 20 minutes to ensure that all dichloromethane has evaporated. Alternatively, heat the liquid until it stops losing mass. Once all
of the dichloromethane has been evaporated, transfer the pure, colorless chlorobenzene to a suitable bottle for storage.
Final product in storage bottle
Discussion of Results
9.98 grams (roughly 9 mL) of pure chlorobenzene were collected. This corresponds to a 40% yield based on aniline. The following factors may be
responsible for this relatively low yield:
• This is a relatively small scale of this reaction. Dealing with such a small amount of material causes more loss during workup (e.g. separatory
funnel washes, transfers between containers, etc.).
• Appreciable loss occurred during steam distillation. The distillation was stopped when distillate began to come over extremely slowly. Perhaps if
the reaction flask was heated more intensely, less steam would have condensed prematurely. This would allow the distillation to be continued longer,
likely yielding more chlorobenzene.
• A small amount of chlorobenzene is lost to evaporation during the removal of dichloromethane.
Attachment: Chlorobenzene from Aniline via the Sandmeyer Reaction-2.pdf (513kB)
This file has been downloaded 2356 times
|
|
|
Bert
Super Administrator
Posts: 2821
Registered: 12-3-2004
Member Is Offline
Mood: " I think we are all going to die. I think that love is an illusion. We are flawed, my darling".
|
|
Nice lab, well documented.
Where can you buy aniline as an amateur chemist in USA... It's another of those materials that seems to have "gone out of fashion".
Rapopart’s Rules for critical commentary:
1. Attempt to re-express your target’s position so clearly, vividly and fairly that your target says: “Thanks, I wish I’d thought of putting it
that way.”
2. List any points of agreement (especially if they are not matters of general or widespread agreement).
3. Mention anything you have learned from your target.
4. Only then are you permitted to say so much as a word of rebuttal or criticism.
Anatol Rapoport was a Russian-born American mathematical psychologist (1911-2007).
|
|
|
ParadoxChem126
Hazard to Others
Posts: 104
Registered: 5-4-2013
Location: USA
Member Is Offline
Mood: No Mood
|
|
Thanks. I acquired my aniline from Elemental Scientific, but you do have to pay for the UPS exemption pack in order to ship it.
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
This is another way to make aniline OTC. I'm planning on trying it, it seems much easier and somewhat safer than benzoate-> benzene->
nitrobenzene-> aniline
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
Quote: Originally posted by Bert  | Nice lab, well documented.
Where can you buy aniline as an amateur chemist in USA... It's another of those materials that seems to have "gone out of fashion".
|
See if you can buy acetanilide instead. You can convert it to aniline in 85+% yield through a facile acid hydrolysis and vacuum distillation. The
upside is that it is a relatively nontoxic solid and as such, shipping is far easier.
Plus, a number of possible transformations require acetanilide anyway and it is stable in storage unlike the free amine.
[Edited on 9-25-14 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
ParadoxChem126, You're 40% yield is not off-base. I met someone who tried the same reaction with similar results. I forget their exact yield I wanna
say it was ~50%. No that's not covert SWIM talk, I wish I had some chlorobenzene or bromobenzene. I'll probably give this reaction a whirl sometime
this year.
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by smaerd | | ParadoxChem126, You're 40% yield is not off-base. I met someone who tried the same reaction with similar results. I forget their exact yield I wanna
say it was ~50%. No that's not covert SWIM talk, I wish I had some chlorobenzene or bromobenzene. I'll probably give this reaction a whirl sometime
this year. |
I can confirm that crap yields seems to be par for the course with Sandmeyers. I got an 40% yield of Bromobenzene from aniline years ago. This was
exceptionally discouraging after my first Sandmeyer with 2-nitro-4-methylaniline smoothly gave 90% yield of the aryl bromide. Turning p-toluidine into
the phenol also gave 35% or so yield.
[Edited on 13-10-2015 by UC235]
|
|
|
ParadoxChem126
Hazard to Others
Posts: 104
Registered: 5-4-2013
Location: USA
Member Is Offline
Mood: No Mood
|
|
How did isolate and purify the product? Did you use steam distillation also?
Steam distillation of small amounts of material seems to lead to considerable loss.
|
|
|
ParadoxChem126
Hazard to Others
Posts: 104
Registered: 5-4-2013
Location: USA
Member Is Offline
Mood: No Mood
|
|
I have put together a video with footage detailing this procedure:
https://www.youtube.com/watch?v=GgsCLoPi24I
|
|
|