Klute
International Hazard

Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
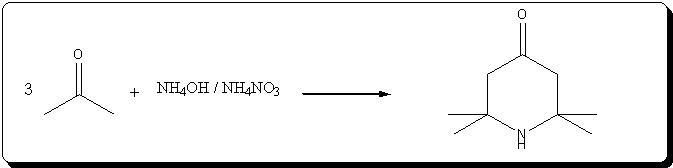
Synthesis of 2,2,6,6-tetramethylpiperidin-4-one (Triacetoneamine)
Over six months ago, I prepared a bottle to make some triacetoneamine, to oxidize it to 4-keto-TEMPO as a catalyst of benzylic/secondary alcohols
oxydations. Initially, I had planned on leaving it for a couple of weeks, but as time passed I forgot about it, reminding myself to distill it every
now and then...
Being very bored because of my incapacity to manipulate after having wounded my left hand, I couldn't resist anymore and decided on distill the
mixture. Someone gave me a hand (both litteraly and figuratively  ) to put the
distillation setups up, and I was off to isolate my 2,2,6,6-tetramethylpiperidin-4-one. ) to put the
distillation setups up, and I was off to isolate my 2,2,6,6-tetramethylpiperidin-4-one.
Synthesis of 2,2,6,6-tetramethylpiperidin-4-one (Triacetoneamine)
Condensation
I didn't take notes when I mixed the reagents, so I'm not 100% sure of the numbers. Keep in mind that this reaction wasn't done to obtain good
yield, but just to obtain some product to work with. A lots of improvements can be done, especially during workup, I just wanted to get my product
with the least of efforts, considering my handicap.
In a 500mL bottle, 200mL technical acetone was mixed with 100mL 20% aqeous NH3. Roughly 5g of ammonium chloride and 5g of silicagel were added, and
the bottle was shaken. This was left at room temp for over 6 months, shaking occasionally. A slight pressure was released a few times. The ammonia
smell was less intense as it was as the beggining.
Picture 1

Distillation of the excess reagents
The now fluo (when a light was directed at it) orange mixture was filtered through a cotton plug into a 500mL distn flask. The liquid had a
viscosity comparable to that of cooking oil. A simple disnt setup was attached, with a 15% H2SO4 wash bottle connected to the vacuum inlet. As heating
started, the air present in the system was chased out, at a flow of 1bubble/1-2sec, which increased as ebullition started. Then ammonia gas was
absorbed, as seen but the instant absorbtion of the bubbles and a "cracking" sound. At first, some condensate started passing under 40°C, surely
acetone vapors pushed by the ammonia gas. Then a constant take off started, with vapors temp slowly increasing from 50°C to 75°C. An orange oil
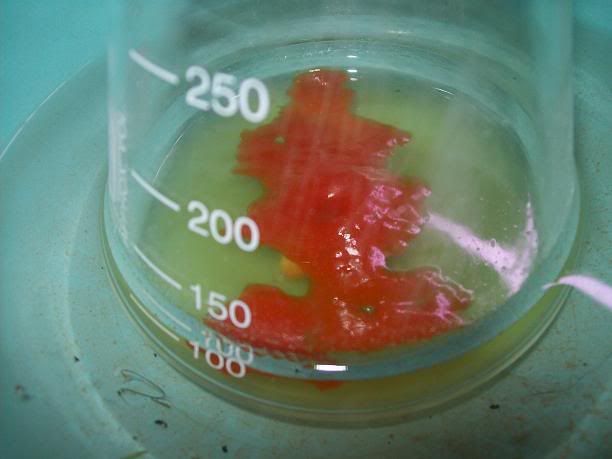
started crashing out as temp increased above 60°C [Picture 2], and some clear organic oil was seen steam distilling (Note 1). At
75°C vapor temp, the flask was cooled, and the top orange oil (Note 2) was seperated in a seperating funnel [Picture 3][Picture
4].
<table><tr><td>Picture 2</td><td>Picture 3</td><td>Picture
4</td></tr><tr><td>  </td><td> </td><td>  </td><td> </td><td>  </td></tr></table> </td></tr></table>
The orangish aq layer was returned to the distn setup, and distn continued until vapors temp increased over 85°C. A very small amount of oil
seperated out. The clear distillate had a small amount of oil, which quickly turned to a white solid (Note 3).
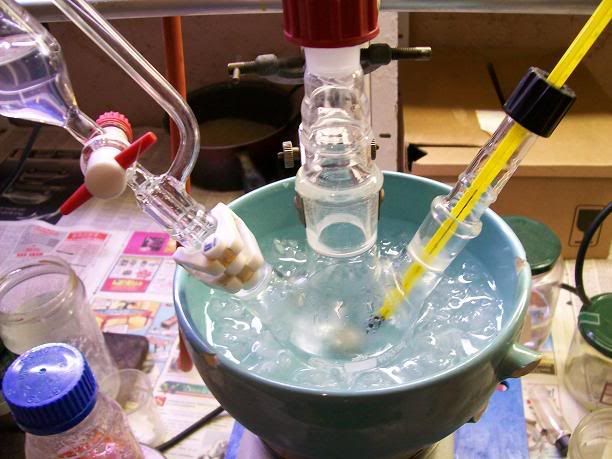
Vacuum fractionnation of the crude oil
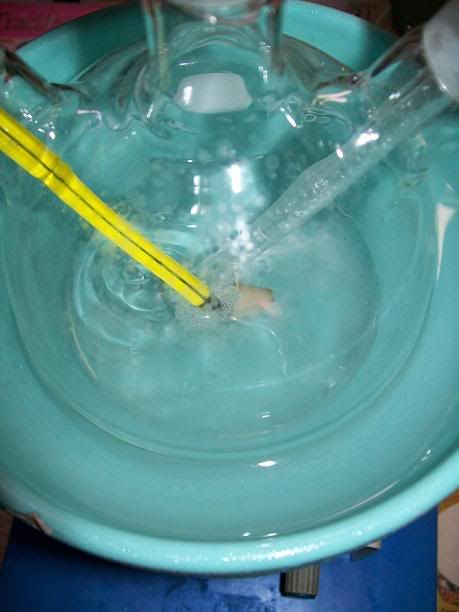
The combined organics were directly introduced into a 250mL distn flask [Picture 5], attached a a vacuum fractionnation setup [Picture
6] , using a apirator as vacuum source.
Picture 5

Picture 6

At first some water passed over, with a clear light yellow oil (Note 4). Some white smoke started passing. Vapor temp stabilized at 55°C.
Roughly 20mL of water were collected, with ~5mL of oil floating above. Heating was increased, more smoke was produced, to quite an extent [Picture
7]. Then very strong foaming started, to a point were vacuum couldn't be left on for more than a few seconds before the foam dashed up the
vigreux. The black/brown residu was transfered to the 500mL flask (heavy smoking in air), and distn started again. This time the foaming was well
contained.
Picture 7

Finally, temp increased, smoking diminished, and a light golden yellow oil started passing over [Picture 8]. A first fraction was collected
(70-95°C) [Picture 9], weighing 10.44g, followed by another one at 95-105°C [Picture 10] weighing 18.5g. The second fraction was
slightly darker. Distn was stopped as head temp increased above 105°C.
Picture 8

Picture 9

Picture 10

Notes:
Note 1: This was surely the triacetoneamine. This means it could be purified by steam distn if someone is willing to take the time..
Note 2: This oil was pretty viscous, as motor oil, and had a sweetish amine smell, pretty consistant with Guy's description.
Note 3: This is surely the product forming a hydrate with the distd water. I wanted to collect the solid, but when cooling a stupidly
forgot to disconnect the wash bottle, and some H2SO4 was sucked back, dissolving the solid.
Note 4: The large amount of water passing could be du to some uncondensed product cyclizating (sp?) and thus generating water. Or it's
just that the oil can dissolve large amounts of water. Solvant extraction and drying could be more efficient and avoid excessive degradation (smoke).
A few attempt of making a salt were made, adding some H2SO4 in abs EtOH (10% w/v) in the oil dissolved in a little IPA caused some white solids to
appeared but they formed an oily layer after a few seconds. Adding 30% HCl in IPA follwed by acetone didn't produce any cristallization. Any
suggestion regarding salt formation are welcome, as i would like to keep some product as a salt for better stability, because I don't want to oxidize
everything in one go.
To oxidize this amine, i will surely try H2O2/NaHCO3, and perhaps Oxone if the results aren't great. Any advise on this subject are also welcome.
Hopefully, a very efficient oxydation catalyst is at the reach of home chemist. This procedure requires long reaction times, though a few weeks are
surely only needed, but using dry NH3 gas, although more involved, cut reaction times to a few hours (see mentionned patents).
The broad uses and numbers of related publications is very inspiring.
I also want to try reducing the keto- to hydroxy- , to hydroxy-TEMPO, but i'm scared oxidation of the amine would also oxidize the hydroxy group.
But things like acetoxy-, methoxy-, etc could be obtained if a easy way of protecting the secondary amine could be found. Any ideas?
Well, perhaps 4-keto-TEMPO is as efficient as the rest, so why bother.
Once the catalyst is ready, the results of the oxidations will be posted in the benzylic/secondary alcohols oxidations thread.
[Edited on 29.10.13 by bfesser]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Oxydation of 2,2,6,6-tetramethylpiperidin-4-one (Triacetoneamine) to 4-Oxo-2,2,6,6-tetramethylpiperidine-N-oxyl (4-oxo-TEMPO) with alkaline
H2O2
The procedure from US 0256312A1 given above was followed, using AcONa as cocatalyst. The workup was inspired by WO8705222.
All the used glassware was washed with distilled water to avoid contamination with metal salts.
18.5g (119.18 mmol) of distilled 2,2,6,6-tetramethylpiperidin-4-one (Note 1) were placed in a 100mL 3-neck RBF with a stir bar[Picture
1]. 5mL of water were used to rinse the container. This formed a homogeneous orange solution.
Picture 1

In a seperate container, 1.05g (12,5mmol; 0.95eq) of NaHCO3 were covered with 5mL of water, and 0.2g (3.33mmol; 0.27eq to the NaHCO3) of acetic acid
were added dropwise, with stirring (CO2 evolution). The erlenmeyer was slightly heated to dissolve the NaHCO3. This solution was added to the RBF,
giving two layers [Picture 2], and heated in a oil bath to 60°C.
Picture 2

17.93mL (208.57 mmol); 1.75eq to the TAA) of 35% H2O2 were then added dropwise over 3h, keeping the temperature between 55 and 65°C. The orange
color gradually turned dark red over the first hour [Picture 3], and darkened further [Picture 4]. At the end of the addition,
stirring was continued for another hour at 60°C [Picture 5].
Picture 3

Picture 4

Picture 5

The flask was cooled to room temp, and 10mL of water was added to the dark red emulsion, followed by 25mL DCM. pH was brought to 2 with 10% HCl (very
slight bubbling), and the two layers seperated. The orange aqueous was extracted with 3*10mL DCM. The combined extracts were washed with 50mL water
Picture 6, and 50mL brine Picture 7, before been dried over Na2SO4 and placed in the freezer.
The solvent will be removed tomorow.
Picture 6

Picture 7

Final extract

Notes:
Note 1 : The initially clear yellow freebase had turned orange when kept over night at room temp.
Comments
This reaction is very smooth and easy to perform. I'm sure the H2O2 addition can be done more quickly (they add it over 4h in the patent). The
demethydioxirane oxidation looks very interesting by the very short reaction time and easy workup, but preparing pur DMD in acetone is tedious. I'm
sure adding oxone to acetone and using the suspension directly could work out nicely, filtering before evaporating the solvent.
I'm not too sure how to purify the product, i don't want to do a coluum, being out of chromatographic silica. Apparently the mp is pretty low
(~30°C), so recrystallization would be tedious. Any ideas?
Can't wait to try the oxidations out! Even though i have too 
[Edited on 6-4-2008 by Klute]
[Edited on 29.10.13 by bfesser]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Synthesis of 4-oxo-TEMPO according to US 3,959,295

first step:
In a 250mL 4-neck RBF, equipped with a gas inlet, a condenser, a thermometer and magnetic stirring, 25mL (340 mmol) of acetone and 9mL of MeOh were
charged, followed by 1.1g (20.56 mmol) of freshly fused NH4Cl.


A 500mL erlenmeyer was charged with ~100g of humid, crude NH4Cl (byproduct from hexamine hydrolysis), containing roughly 80g (1.5 mol) of NH4Cl,
and a stir bar. The erlenmeyer was fitted with a 250mL addition funnel, containing 150mL of 40% w/v NaOH solution (1.5 mol). A gas outlet was
connected to the top of the addition funnel, linked to a wash bottle containing solid NaOH pebbles, itself connected to the gas inlet of the RBF.

The NaOH solution was slowly dripped onto the NH4Cl, generating a constant, easily monitered flow of NH3. the acetone/MeOH was gassed at
1bubbles/sec, with vigorous stirring, at room temp (17°C). A latex glove was attached to the top of the condenser, keeping in any unabosrbed NH3. It
was not "blown up".
Gassing was continued for 3H, until the mixture was saturated. Near the end, the temp rosed over 30°C, so an ice bath was applied to keep temp
under 15°C. Once saturation was definatively attained, the gas inlet was removed.
Second step
75ml (1.02 mol) of acetone are then added, turning the whole mixture milky white as a fine solid precipitates, and agglomerates upon stirring.


The flask is then heated to 50°c with an oil bath. After a few minutes, the solid dissolves completly, offering a water-clear solution. After1 hours
stirring, the mixtures takes on a yellow colour, and afetr 15H a dark red solution is obtained.


The condenser is replaced by a fractionnal distn setup, with a 150mm vigreux, and the gas tube connect to the vacuum inlet and immersed in a dilute
H2So4 solution. Heating is increased, and the excess acetone removed. After ~75mL are distd, slight vacuum is applied.

A dark red, viscous residu is obtained. It fumes when vacuum is disconnceted.

2.25mL (125 mmol) of water are added to the cooled residu, and the flask is immersed in a ice water bath with slow stirring. After 1 hour, there still
isn't any solid forming. apparently the product is too impure to be able too form a hydrate directly.
The residu is then titrated with conc. HCl. Over 20mL are required until acidic to dampened pH-paper. 100mL of IPA are added, and distn under vacuum
at 60°C. this is renewed with 50mL IPA. Unfortunaly, the distn was left unattended for too long, and the residu heated up considerable when the IPA
was completly removed, but not for long.
The thick black/red syrup was cooled down, thickening alot, and 50mL toluene added. A semi-solid tarry goop is formed, which doesn't solidify when a
little sample is triturated under acetone, although it looks likes a cristalline paste, it stays liquid.
30mL acetone are added to the two layers, the solvent top layer taking ona red color, without much effect on the tar. 30mL of IPA are added, which
dissolves most of the tar, forming a black/brown homogeneous layer, and a white/yellow solid sperates out immediatly. This is left to sit for 30min,
then vacuum filtered. A yellowish powder is obtained, which is generously washed with acetone, and pet ether before being dried by suction, giving a
very light, fluffy beige poxder.


A total of 10.7g (55.87 mmol) of triacetoneamine hydrochloride is obtained. The solid will be recrystallized with the old sample, from IPA/acetone,
or IPA/pet ether.

Comments
The workup is the problem here. The cristallization is far from optimal. The filtrate has been placed in the freezer to see if more product will
cristallize, but next time I will rather try adding conc. H2SO4 to the reaction medium directly as advised by the patent, or adding ethanolic oxalic
acid to the residu, as advised in some of the articles KMnO4 very kindly posted.
The oxalate salt seems pretty easily isolated, and fairly stable.
It's a pity the hydrate didn't readibly form, this would definatively be th ebest way of stocking TAA for short to medium periods. No need of
basifying before oxydation. I guess it could be kept in the fridge for longer stocking time, but as it is commercially avaible under this form, I
guess it's pretty stable.
In any case, this seems to be a good reaction: no need of gassing for several consecutive days, waiting a week, etc. Apparently the methanol does a
good job at dissolving alto of NH3, as pur acetone doesn't dissolve much by itself, hence the consecutive gassings in sevral articles. The gassing is
pretty staright forward and doesn't require constant presence once stable. Just gas the shit out of it until saturated . I think a little less acetone could be added in the second step, I followed the
second series of examples in the patent, but the first one used 1/3 the amount of acetone in the second step.
Considering the ease of the reaction it would be adviseable to do it at a bigger scale. If the TAA can be directly isolated from the reaction medium
by conc. H2SO4 as claimed by the patent, no need of distilling the excess acetone, which saves quite some time. The ~8.6g of freebase should yield
more than enough 4-oxo-TEMPO, but I might just try another reaction with using either H2SO4 or oxalic acid to isolate the amine.
Once thta's done, oxidation time!
Come people, jump in! This is easy and rather a break through in home-chemistry: a 100$/gram catalyst ( !! ) easily synthesized from scratch!
The second part, checking out the efficienty and range of use of this catalyst is going to be a pretty large project too.... Obviously, i will be
starting with benzylic alcohols to aldehdyes oxidations, becasue that's why I decided on try to make the catalyst in the first place, but it can also
be used for aliphatic aldehdyes, nitriles from amines, etc
Here is the patent for the lazy people  : :
[Edited on 23-5-2008 by Klute]
Attachment: US 3959295.pdf (264kB)
This file has been downloaded 977 times
[Edited on 29.10.13 by bfesser]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Oxydation of triacetoneamine with H2O2/K2CO3
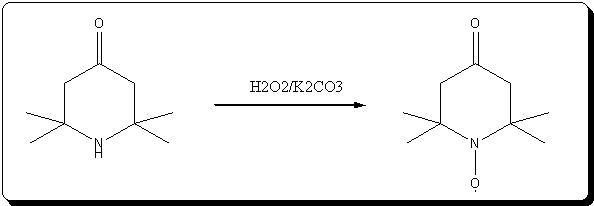
This is a small variation of the methode described in the article KMnO4 retreived lately, that i attached just above.
A made a few modifications, and could have done things a little better; but at least know I know what I will do next time :)
In the procedure, they use anhydrous Na2CO3. At home, I had the choice between very technical (hardware garde) hydrated Na2CO3 (unknown hydratation,
less than decahydrate, and of questionnable purity, possibly containing soaps/detergents), and anhydrous 98% K2CO3, so I prefered using K2CO3. It
should be noted that in most procedures using H2O2/CO3 2-, the sodium counter ion is used. In the patents, they claim potassium carbonate can used,
and a example gives a very high selectivity, so it shouldn't be a problem.
Procedure
In a 250mL wide neck erlenmeyer, 21.8g (157.73 mmol, ~3.4eq) were dissolved in 64.35mL (631.02 mmol; 13.7 eq), 30% H2O2. This caused some warming up
and fizzing. The milky solution was cooled in a ice bath with slow magnetic stirring.

8.83g (46.12 mmol) of recrystallized TAA.HCl were crushed into a finer powder, and added in portions to the solution. AT first, nothing much seemed
to happen, no CO2 evolution, the powder jsut formed a suspension.

1.88g (47,00 mmol) of Naoh were weighed and added as a solid in small portions to the suspension. The solids seemed to increase in volume, and
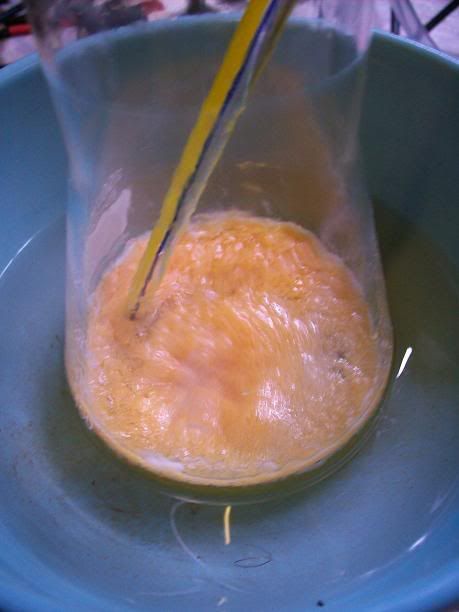
whiten. A thick slurry of a milky white fluffy solid was formed: the TAA formed a hydrate as soon as it was based. As O2 bubbles were evolved, the
voluminous solid formed a foam, hard to stir in surface. It was broken down regularily with a thermometer, to facilitate magnetic stirring.



A limpid colorless liquid was under the layer of foam, looking alot like egg's white that has been "beaten" to snow (don't know the expression in
english, you knwo what I mean :) ). It was continuously stirred with the thermometer to mix up the foam.
the temperature was left to climb up to 20-25°C, sometimes up to 30°C, but kept under that temperature by applyling a ice bath now and then. There
wasn't any strong exothermic reaction, the heating was due in part to the hotplate used to stir. There was continuous, but gentle, evolution of
bubbles.
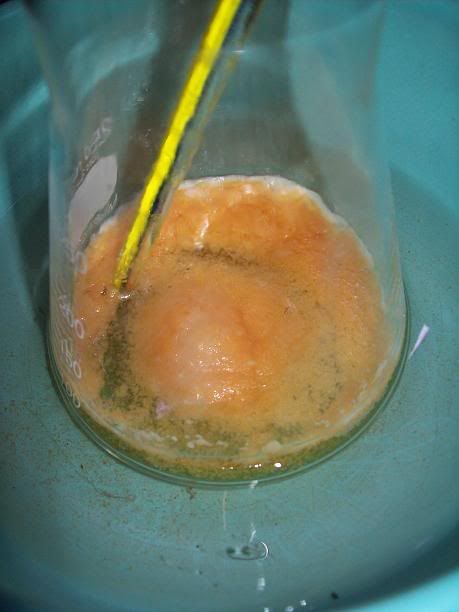
After 4h, the liquid started to turn limpid yellow, and the amount of solid/foam seemed to diminish. Stirring was much more easier.



At 6H total reaction time, the white foam had turned to light orange cristallin solids floating on the surafce of the liquid; the temperature ahd
increased over 40°C at one point, and was quickly cooled donw to 15°C with the ice bath. There was still noticeable bubbling.

After 20H, a dark orange/red semi-solid tar like gum is obtained. No more bubbling can be seen.
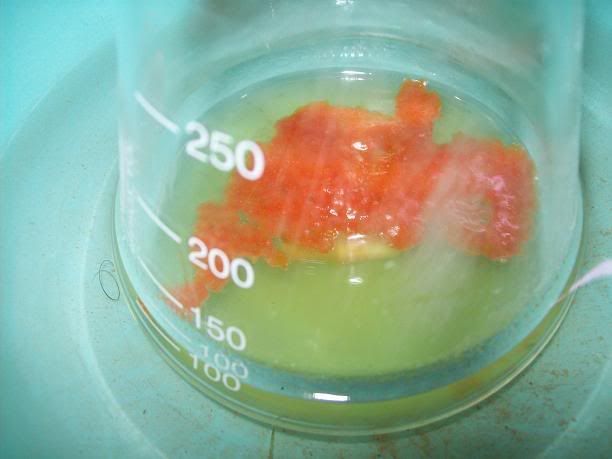
I'm thinking starting the work up tonight, as it doesn't seem to evolve that much now. I hope the semi-solid will solidify entirely whne refrigerated,
to be able to filter it effectively, otherwise I will have to do a solvent extraction. In that case, I will neutralize with acetic acid until I have a
pH of ~4, so that any unrecated TAA will stay in solution and the nitrosyl radical will not be degraded.
Comments
All the glassware was washed with demineralized water to avoid contamination with metal salts, which would promote the decomposition of the H2O2 (note
the huge excess).
The reagents were added in a bad order: the H2O2 was added to the weighed K2CO3, which caused some lumps at the begginign, and possibly a little
decomposition from the basic conditions when only the first portion of H2O2 was added.
Apparently, potassium carbonate cannot freebase TAA.HCl. I will have the double check the pKa of TAA, but I don't recall it was that high... But, I
wasn'texpecting the formation of the hydrate, so the transition could be hard to notice with the carbonate.. If the amine was already freeabsed,
adding the NaOh would cause extra decomposition of the H2O2, as it is favored by high pH.
I should have: added stoechiometric amount of NaOH to TAA.HCl in minium water, then added the K2CO3/H2O2 solution to the freebase/hydrate (prepared
by adding the K2CO3 gradaully to the stirred and cooled H2O2). I think it is important to cool the recation at the beggining of the addition, as the
reaction could spark off and over heat if not controlled.
I'm not sure if the hydrate formation can be avoided if using not-so-cold temperatures, or adding the freebase to a concentrated carbonate solution.
Even if it isn't practical, it doesn't pose a big problem to stir, it's just that the layer of foam isn't well mixed with the aqueous if not stirred
by a rod to send it into the vortex.
Next time, i might try the bicarbonate procedure again, but use their workup which seems to be a breeze compared to extraction, washing evaporation
and recristn.
PS:
from US 5,431,901:
| Quote: |
A. 2,2,6,6-Tetraperdeuteromethyl-4-oxo-3,5tetradeuteropiperidine (Triacetoneamine-d16) (V)
A mixture of ammonium-d4 chloride (3.45 g, 0.06 mole), acetone-d6 (99.5 atom %D, 12.5 ml, 0.15 mole), anhydrous sodium carbonate (3.18 g, 0.03 mole)
and magnesium oxide (3.0 g) was added to a 250 ml round-bottomed flask. The flask was capped with a rubber septum and wired, then the reaction mixture
heated in an oil-bath at 50° C. for 3 days. After cooling, 20 ml of acetone was added to the reaction mixture and the resulting mixture was filtered.
The recovered solid was crushed into powder, washed with 15 ml of acetone and then filtered with suction filtration. The combined filtrates were
concentrated to dryness. The resulting red liquid (7.2516 g) was distilled under reduced pressure to obtain 4.7480 g (56.7%) of a bright yellow liquid
(b.p. (boiling point) 54°-55° C./1.9 mm Hg) that solidified when chilled in a dry ice/acetone bath. The solid product subsequently was used without
further purification. Recrystallization of an analytical sample from anhydrous diethyl ether yielded white crystals, mp. 57°-58° C. [lit. 58° C.];
IR(KBr, cm-1):3580(m), 3260(m), 2220(m), 1700(s), 1530(w), 1265(s), 1140(m), 1050(m), 930(w); 13 C-NMR (CDCl3): 31.03(m), 53.50(m), 54.88(s), 211.19
(s).
|
I guess this is basicly the procedure referenced in the zeolite absorption article, but using d-ammonium chloride instead of 15-ammonium sulfate....
No gaseous ammonia, or extended periods (well 3 days, but better than 3 month :) ). i will definatively try this out.
[Edited on 28-5-2008 by Klute]
[Edited on 28-5-2008 by Klute]
[Edited on 28-5-2008 by Klute]
[Edited on 29.10.13 by bfesser]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Synthesis of 2,2,6,6-tetramethylpiperidin-4-one according to US 3,959,295, second try
This time I (nearly) followed the patent's procedure, and it's proposed workup with conc. H2SO4. Works like a charm!
First Step
In a 500mL 3-neck RBF, equiped with a condenser fitted with a latex glove fixed with an elastic, a gas inlet, a thermometer and magnetic stirring,
63.2mL (1.377 mol) of technical acetone and 15mL of MeOH were introduced, followed by 2.2g (41.12 mmol) of freshly recrystallized and dried NH4Cl and
1g CaCl2 pellets.
Roughly 150g (2.8 mol) of freshly recrystallized NH4Cl were charged in a 500mL flat bottom flask, with a stir bar. The flask was mounted with a 250mL
addition funnel, itself mounted to a gas outlet, connected to the gas inlet of the RBF in series with a solid NaOH washbottle.
A solution of 120g (3 mol) NaOH in 250mL dH2O was charged in the addtion funnel



The NaOH dripped onto the the NH4Cl, producing a slow, constant flow of NH3 gas. The RBF was immersed in a cold water bath, to which ice was
periodically added in order to keep the internal temp under 20°C, most of the time between 10 and 15°C.
the flat bottom flask was heated on a hot plate to acheive better desorption of the NH3.
Once all the NaOH solution had been added, the NH4Cl and CaCl2 had completly dissolved into the colorless solution.
Increasing the heating of the flat bottom flask generated increasingly more NH3. Once the solution was nearly saturated (the glove started to get
blown up), 50mL of technical acetone were added to the flask's contents. The initially colorless solution immediatly turned milky white, and gradaully
cleared up as a white solid agglomerated onto the sides of the flask.


Gassing was continued as long as NH3 was generated. A very rapid flow of gas was now produced by the nearly boiling solution/slurry.
(Remark: In the first try, I dismantled the gas generator shortly after all the NaOH had been added. Apparently, a very large amount of ammonia
stays into the NaCl slurry, and extended heating is required for complete desorption. This must surely account for the low yield of the first
try.)
Once the flow of ammonia produced came to a near halt, the gas inlet was disconnected and immersed in a dilute H2SO4 solution. There was no suck back
at all.
The gassing took a total of 7H to proceed entirely. This period could be shortened by increasing the flow at the beggining of the gassing.
The solution was bubbling from ammonia (saturated) as it heated back up to 25°C (ambient), so a few tiny holes were put through the latex glove to
avoid excessive overpressure.

92.2mL of technical acetone were added (total amount of acetone introduced: 63.2 + 50 + 92.2 = 205.4mL; 4.477mol), without apparation of any
white solid, and another 1.1g (20.56 mmol) of NH4Cl and 1g of CaCl2 were added. The ammonium chloride dissolved immediatly, the CaCl2 did so more
slowly.

The colorless solution was stirred for 2h at room temp to avoid wastefull NH3 release. After that period, a small aqueous layer was noticed at the
bottom of the flask.

Second Step
The flask was then heated to 55°C in a oil bath. AS the flask heated up, there was only minimal NH3 release.
After 10H of heating, the solution had taken a light yellow colour.

Following the patent's procedure, 14g of H2So4 were charged into a addition funnel, and slow addition started. There was a extremly vigorous reaction
as the first drop touched the surface, generating alot of white fog and a strong sizzling noise.


The acid on the sides of the flask quickly turned to a white solid as it reacted with ammonia. White solids started appearing in the solution. The
addition was stopped, and it was decided to continue reflux without addition of acid.
heating was continued for another 5h, at which point the solution had taken a blood red colour.

The flask was cooled with a cold water bath, and maintained in a ice bath while conc. H2SO4 was slwoly dripped in. Again, the reaction was vigorous,
producing lots of white fog.

As the addition proceeded, the reaction became less intense, aswell as the generation of the fog. Any acid on the sides of the flask didn't not turn
to a solid as previously even after prolonged exposure.
The temperature was kept under 35°C by changing the ice bath regularly. 75mL IPA was added to thin up the slurry and help stirring. A thick red
slurry was formed. Once pH was acidic (to dampened universal pH paper, ~5), acid additon was stopped (over 25mL were required), and stirring at 10°C
was continued for 45min. The pH was checked at the end o fthe stirring period, and was found to still be acidic.
The thick slurry was vacuum filtered on a large buchner, taking some time to pass (the filtrate started boiling sevral times). The light bordeau cake
was washed with 200mL acetone, 75mL 2:1 IPA:Acetone, then 100mL pet ether (filtering was much easier after the first wash). This gave a off white
solid cake, and a blood red filtrate.


The filtrate placed in the freezer after been sealed with celophane, and the solid was dried by suction for 20min, then under a lamp.
73g of a light, free flowing off-white/beige powder was obtained.


The powder will be extracted/recrysatllized from methanol to remove ammonium sulfate and other impurities. It has a light smell of the free amine.
Comments
What a releif! the workup is definatively much simplier than assumed, and avoids lengthy and destructive distillation of excess acetone and added IPA.
The yield is much better this time, surely because I took time to completly deplete the NH3 generator. Last try I must have wasted at least half the
NH4Cl this way, as alot of gas remains in the slurry. Next time, I might consider simply heating 20% ammonia to generate the gas, as I have no more
NH4Cl left.
the gradually addition of the acid during the reflux just didn't inspire me. I was scared of neutralizing too much unreacted NH3, and end up with a
bunch of ammonium sulfate. Considering that the H2SO4 didn't immediatly react with the atmospher inside the flask as it was introduced during the
titration, i think it was a good idea.
I added a extra portion of acetone before finishing the gassing, as the solution was getting saturated, and I didn't want to waste any NH3. Adding a
little more methanol could have helped.
Hopefully the product isn't too contaminated with ammonium sulfate, and a simple MeOH extraction and subsequent recrysatllization will be enough. A mp
determination is in order, though I have yet to find the litterature value for triacetoneamine hydrogenosulfate.
Conclusion: I would really recommend this recation to anyone desirable to obtaining a fair amount of TAA. The gassing stage isn't such as hassle, as
it it virtually takes care of itself, and the workup with H2SO4 (or ethanolic oxalic acid) is very easy and requires very little work. The gassing
time can be shortened, it just that I went too slow at first; I'm impressed at how well the mixture absorbs the ammonia, even with a flow of bubbles
close to 3-4 bubbles/sec, the glove didn't get blown up entirely until the end were the solution was saturated. It was pretty impressive to see the
massive bubbling of the solution from NH3 release once gassing was stopped.
Side note: I have tried the procedure without gaseous ammonia I posted a few post back, using NH4Cl, K2CO3, MgCO3 and CaO (I didn't have any MgO at
hand, and was unsure about using either MgCO3 or CaO, so just tossed both in) into a sealed bottle kept at ~40°C. In one day it has already turned
blood red, and the solids have turned from very fine powders to a spongeous mush. I will update with the results once workep up.
[Edited on 1-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
MeOH extraction of the above crude bisulfate was tedious, so it was decided to purify the TAA via it's hydrate.
The MeOH-wet powder was dissolved in 300mL warm water, resulting in a yellow suspension of a fine white/beige solid, which was vacuum filtered
(calcium sulfate) and washed with a little dH2O.
The yellow solution was basified with conc. NaOH until pH>13, at which point the solution became cloudy, and smelled of ammonia. It was placed in
the fridge, then in the freezer for 45min.
the mixture seemed to be entirely frozen. It was left to warm up for 15min, then vacuum filtered. It had turned to a gelatinous mass, which was long
to filter. Gelatinous scales slowly collected.


After testing their insolubility in acetone, the gel-like cake was flooded and triturated under 150mL acetone for 5min, vacuum disconnected, and
vacuum filtered again. Beautifull white scales were collected, pressed down and dried by suction for 30min, then layed down under a lamp to air dry.


After 4h, the total weight of pur 2,2,6,6-tetramethyl-4-oxopiperidine hydrate was : 62.0g. 
EDIT: a second crop of the hydrate was obtained by adding K2CO3 to the filtrate and placing it in the freezer. The white crystals formed were vacuum
filtered, and washed with acetone before being dried by suction and under a lamp. The solid is much more "soggy" and is possibly contaminated by
K2CO3, although it melts around 50-60°C.
I'm unsure of which solvent to use to recrysatllize it. I think I recall undried ether been mentionned somewhere, but can't find the mention again.
What could I use, water? It might not be the most effective way to remove K2CO3..
Well, next step is NaBH4 reduction! I now have some TAA hydrate and recrystallized bisulfate (from the MeOH cristn) that will be properly
characterized.
BTW, can anyone suggest a easy and good yielding method of reducing oximes? I was thinking of doing a CTH to reduce the oxime of 4-oxo-TEMPO or TAA
(depending on reducer used, hydrogenation reduces the nitrosyl radical IIRC) as I've got some experience in the field, but if I could find a another
good reduction that doesn't involve Pd/C (or messy zinc), I would be happy. I have heard one unreferenced proposition of using NaBH4 alone, but
haven't found much info so I doubt it a bit. Any suggestions welcome.
Still the crude crop to purify and the unrecrystallized TAA.HSO4 to add.
[Edited on 3-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Thank you very much Guy
Apparently when purified via a salt, the TAA anhydrous freebase is pure enough to crisatllize at room temp, and forms beautfillu small white needle!
The problem is the mp is slightly depressed and in the day when it's hot they completly melt to a yellow oil, and crisatllize back at night
The hydrate stays as is though, and seems like the ideal way of storing TAA, no need of basifying for any further reactions, and if the nahydrous form
is needed, a reflux in toluene with a dean stark or similar should be just enough!
Gaseous ammonia is definatively the way to go!
EDIT: after some though, I'ev decided on posting all my TEMPO-related work in this thread, instead of making a thread for each compound, to regroupe
all the informations on theses derivatives in one big thread.
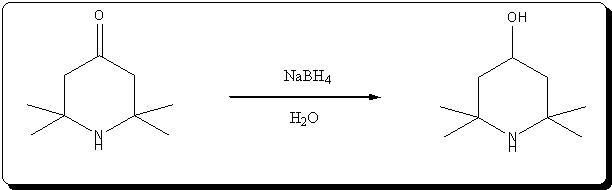
Synthesis of 4-hydroxy-2,2,6,6-tetramethylpiperidine by NaBH4 reduction of triacetoneamine
Ref:
Effect of electrostatic factors on the stereochemistry of the hydride reduction of ketones of the piperidine series
É. A. Mistryukov
Russian Chemical Bulletin (14)10; 1788-1792 (1965)
Very kindly provided by Solo in the ref forum.
Ina 100mL 3-neck RBF, equipped with a condenser, a addition funnel, a thermometer and magnetic stirring, 4.33g (25,00 mmol) of
2,2,6,6-tetramethyl-4-oxopiperidine mono-hydrate (preparation detailed a few posts above) were dissolved in 32ml of dH2O. the dissolution took a few
minutes and was endothermique. The solution was then cooled down to under 5°C in a ice bath.



A solution of 0.53g (14.03 mmol, 2.24 hydride equivalents) NaBH4 in 13mL dH2O containing a few mg of NaOH (to stabilize the hydride) was prepared (a
little fizzing at first, that quickly stopped) and charged in the addition funnel. With steady stirring, the hydride was added dropwise over 10min to
the cooled TAA solution. There was no noticeable appearance change, or visible bubbling. The addition funnel was rinsed with a few mL of MeOH, which
caused some TAA hydrate to crash out of solution. A little dH2O was added until complete dissolution. Once the addition was finished, the flask was
stirred in the ice bath for another 10min, then at RT (15°C) for 2h30.


5mL of 31% HCl were then charged in the addition funnel, and added dropwise, evry slwoly at first as each drop caused massive H2 evolution. The flask
was immersed in a cold water bath, and the temperature kept under 20°C. After ~3mL of acid ahd been added, the gas evolution calmed down at each drop
then ceased completly. The rest of the acid was added in one portion. The completly colorless solution had a pH under 5 (universal pH paper), and had
a smell pretty similar to that of TAA freebase, but noticeably different.

The solution was transfered in a 100mL single neck RBF, and place din the fridge awaiting vacuum distn of half the water tomorow.
Update will be posted when I get time to, possibly not until a day or two (that's why I preferred starting now, pretty busy at work at the moment).
The reaction looks very easy and manageable. Hopefully removal of most the water could be avoided by using more concentrated solutions (or adding
solid NaBH4) and saturating with NaCl or K2CO3, but for the first time I'm sticking to the article by the letter (well nearly ).
Direct oxidation to the nitrosyl in a one pot reaction could be possible, I don't think the borate salts can cause problems, actually perborate
formed by action of H2O2 on borax could be a good oxidant for this type of reaction!
It would just take neutralizing the excess hydride with hydrochloric or acetic acid (directly generating acetate co-catalyst in situ), adding
(bi)carbonate, and slowly adding H2O2 at 50°C.. Both reactions could be done pretty quickly, the reduction is surely finished in less than a hour
(TLC could confirm that), and the oxidation hardly taking more time than that. Place in the fridge, filter the next evening, recrysatllize et voilà!
Let's cross fingers.
[Edited on 4-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I used 5% AcOEt in toluene at the beggining, to elute most of the blue fraction, than gradually came up to 20% AcOEt in toluene to elute the
4-oxo-TEMPO: worked like a charm, and offered a very good seperation (there was a blank fration, no UV-visible compounds for ~50mL between the blue
and orange fraction, using 5uL samples on the plate (twice the amount usually used, most of the time hardly 0.5uL is needed to get a spot with 1%
solutions).
The solvent was removed under vacuum, to give a red oil that crisatllized once excess toluene had evaporated off at RT to give a low-melting red
solid. It will be recrysatllized with pet ether to try and get a sharper mp.
Pictures of the column:






First blue fraction:

it's surely one of the two unsaturated nitro ring-opening compounds mentionned in the article KMnO4 posted and out of which I used the procedure for
the oxydation. They claim ring-opening can happen on workup of the product, the ethyl acetate was removed at atm pressure and contained some acetic
acid. It seems that's all it takes. I will remove the solvent latter on and see which compoud it is (one is an oil, the other a crisatllin substance)
The orange fraction (pur by TLC) once solvent is removed under vacuum.

Picture of the pur product will arrive when done.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
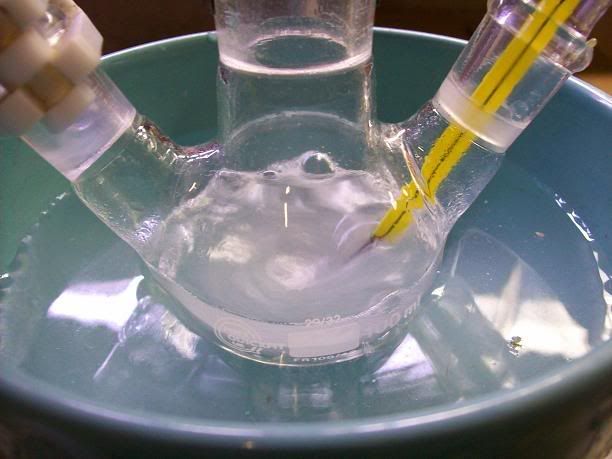
Preparation of triacetoneamine according to US 6,646,127
The patent's procedure in example 22 was followed, using CaCl2 as catalyst.
NH4NO3 was prepared by reacting 53% HNO3 with 30% NH4OH (very vigorous reaction), evaporating the water and drying the salt by melting it.
Acetone was technical grade, CaCl2 pellets were powdered prior to addition.
Procedure
In a 500mL 3-neck RBF, 5g (45mmol) of powdered CaCl2 and 40g ( 0.5 mol) of dry NH4NO3 wee suspended in 220mL acetone. 31mL (0.5 mol) of 30% NH4OH
(Panreac) were charged in the addition funnel, and slowly added dropwise, over 20min.


At first there was a little caking of the solids, then gradual dissolution. The flask warmed up to 40-45°C, so an ice bath was applied to keept temp
under 30°C. Once the addition was finished, most of the solids had dissolved to give a limpid solution.

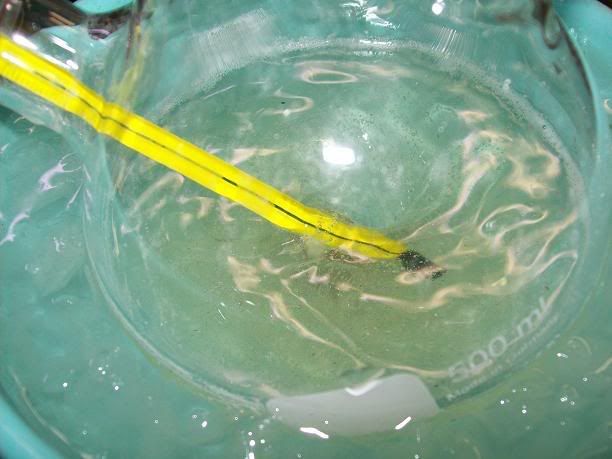

The reaction medium was then stirred at RT for 7H total. The slightly yellow solution gradually turned orange then red, with that caracetistic
"fluorescent" red tint near the end.






The condenser was then replaced by a fractional distn setup, with a short (200mm vigreux column). The acetone was removed under slight vacuum at 45°C
over a 80°C oil bath. the reaction mixture had a strong smell of TAA. The colorless distillate smelled strongly of ammonia. Distn was continued
until no more acetone was distilled, and water started to take-off.n A dark red residu remained.


After cooling down to 30°C, the very viscous blood red residu whic smelled strongly of TAA was transfered to a 1L seperating funnel, and extarcted
with 4*50mL toluene. The extarcts came out golden orange, leaving the blood red color in the viscous aq. layer. After the 4 extraction, the aq. didn't
really smell as TAA anymore, but more as ammonia.


The toluene extracts were combined, washed with 50mL conc. K2CO3, and brine, thend ried over K2CO3 in a stoppered erlenmyer for 20min. The extract was
then transfered to a 500mL beaker, and acidified with 20% v/v H2SO4 in IPA. At first, a white precipitate formed, but as acidification continued, a
dark brown tar_like resin preicpitated also, very similar to the other TAA workups.

There was still a certain amount of white precipitate in suspension. After standing for 5min, most of the suspension was vacuum filtered, leaving
most of the resin in the beaker. 100mL acetone:IPA 50:50 were added, and the resin triturated, dissolving most of theresin and freeing more clear
solid. The suspension was vacuum filtered, thoroughly triturated on the buchner with 50mL acetone, and 50mL acetone:pet ether 50:50 before drying by
suction for 5min. The fine beige precipitate was then placed in a glass cup and dried under a lamp to afford 10.81g (41mmol) of crude
2,2,6,6-tetramethylpiperidone hydrosulfate.

The aqueous layer was re-extracted with 4*50mL AcOEt. These extracts took on a much redder colour. The combined extarcts were washed with 50mL co,nc.
K2CO3, brine, and dried over Na2SO4.


This extract was also acidified until acidic on dampened pH-paper with the same H2SO4/IPA, giving a very fien white precipitate at first, then a
brown/purple voluminous solid. This solid was much harder to correctly clean, giving a sticky paste, and required larger amounts of acetone, IPA and
AcOEt to get it relatively clean. The sticky paste finally dried, giving a crunchy brown solid (4.2g).

The two batches will be recrysatllied from MeOH/pet ether or MeOH/AcOEt.
Comments
Thsi reaction is pretty simple compared to the use of gaseous NH3, but the claime dyields are still not attained. Toluene seems to be a much more
selective solvent for the extarction than AcOEt is, giving a much cleaner product. The workup still needs optimizing though. Direct fractionnation
isn't possibble becasue of the large amounts of inorganic solids present in the syrup. Using diethyl ether as in the patent's procedure, or DCM wasn't
possible because of the ambient temp >35°C here (hot...). Tolueneseems pretty eefctive, but still requires large amounts of solvent. I guess a
gradaully distn of the solvent, and extarction with the recycled toluene could be the best option, as the extract seems pretty clean. Fractionnation
of the TAA freebase might be a better option than direct acidification, it is pretty clear there is a resinous product appering near the end of the
acidification, covering the TAA.HSO4.
Forming the hydrate or oxalate after fractionnation should give a pure product, stable as is. I will try another variation of the reaction, possibly
letting it heat up during the addition s is mentionned in the patent. I'm sure the yield is good, and that the isolation of the product is problematic
(TAA freebase is pretty soluble in water).
I think we are in the good direction though. Using only ammonia could be more practical, or at least catalytic amount s of ammonium nitrate (apart
from the extarction, producing dry NH4NO3 is the lengthy part).
I still have some NH4NO3 left, so will try another recation with a smaller NH4NO3/NH4OH ratio, but same acetone/ammonia donor ratio.
According to the patent, excellent selectivities to TAA are possible at RT, the acetone/ammonia ratio seems to be more important than the temp
regarding acetonine/DAA formation.
[Edited on 23-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
bfesser
|
Thread Pruned
28-10-2013 at 18:01 |
bfesser
|
Thread Closed
28-10-2013 at 18:13 |
|








