| Pages:
1
2 |
golfpro
Banned
Posts: 179
Registered: 18-5-2013
Member Is Offline
Mood: Cap Sensitive
|
|
Who here is capable of Non-Peroxide initiators?
Just wondering how many on this website make/use initiators other than HMTD or AP? I know Ral123 uses AP as initiators still sometimes even though he
can make Lead Azide?? Also say whether you live in the US or not. I am really running in circles trying to think of an initiator I could use that is
substantially better than AP. The first one is Silver Accetylide, that would run me about 80$ and then I need the calcium carbide and the set-up, but
I have read this is not much better than HMTD.. Lead Azide would be close to impossible and to even get NaN3 in the first place, it'd have to be made
w/ hydrazine.
HMTD is so very widely used by hobbyists, and the only accident I have seen of on the internet is the guy w/ the pill of hmtd exploding for no reason.
If the procedure from COPAE is used is it still SUPER dangerous and more likely than not going to explode at the touch? I can't think that you'd need
more than 300mg of it in a straw to initiate ETN which would be all I plan on doing for now.
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!

Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
What about lead or mercury fulminate?
you would have to make it yourself
The procedure is very messy:
| Quote: | Prepare a solution of 1500 mL of 28-29% ammonium hydroxide, 900 mL of water, 375mL of 10% gelatin solution, and 1200 mL of normal sodium hypochlorite
solution. It is absolutely imperative to use distilled water, the presence of any contaminant ions will screw up this reaction! It is possible to use
starch, glue, or glycerol instead of gelatin, but they are inferior. Mix these chemicals in a large glass dish, like a pie plate or bowl, or just use
several portions, as this is nearly a gallon of liquid. This mixture is heated as rapidly as possible and boiled down to one-third of its original
volume. The solution is then cooled thoroughly with ice and suction filtered twice to remove any impurities. When filtering, first use towels (like a
washcloth), then use regular filter paper on top of some cloth (like from a T-shirt).
The resulting liquid is dilute hydrazine hydrate. To make concentrated hydrazine hydrate, mix 144 mL of dilute hydrazine with 230 mL of xylene in a
round-bottomed 500-mL Florence flask. Fractionally distill the mixture in an atmosphere of nitrogen, the xylene will first pass over with most of the
water, then the hydrazine will pass over. Keep the fractions separate of course. The resulting hydrazine hydrate will be 90-95% hydrazine. This
concentration procedure is meant for 60% hydrazine hydrate, since the hydrazine hydrate prepared above may be greater or less than 60%, some
experimentation may be needed to find the proper amount of xylene to use (more xylene is needed for dilute hydrazine, less for more concentrated
hydrazine). To obtain anhydrous hydrazine, mix 20 g of potassium hydroxide per 100 g of >90% hydrazine hydrate in a beaker, let this mixture stand
overnight so much of the water can be withdrawn.
After standing, filter the solution to remove the hydroxide. Add to the filtered liquid an equal amount by weight of sodium hydroxide. Place this
mixture in a round-bottomed 500-mL Florence flask, reflux for 2 hours, then distill in a slow stream of nitrogen. You must use nitrogen or pure CO2,
distillation in air may lead to an explosion! Solutions of hydrazine hydrate can give off extremely toxic vapor. Anhydrous hydrazine is especially
volatile. Salts of hydrazine are much less dangerous, but can give off toxic vapor if they react with a base. Hydrazine sulfate is the least poisonous
form.
|
If you have the hydrazine, might as well prepare NHN, a safer primary that is not very sensitive to physical impact, but is still easily initiated by
flame or electric ignition,
| Quote: | An aqueous solution of nickel nitrate was prepared, containing 8% Ni(NO3)2 by weight. 50mL of the solution was poured into a steel container, which
was then heated to 65 °C. Separately, 100mL of distilled water was warmed and maintained at around 60 °C. Gradually over the period of 30 minutes,
7cm3 of hydrazine sulfate was added into the steel container, simultaneously together with 50mL of the water that had been separately prepared, the
remaining water was discarded. The hydrazine sulfate used was somewhat wet to begin with. The color of the reactants in the steel container changed
from a bluish tint to purple over the course of the reaction. The reaction was stirred for an addition 10 minutes, maintaining the temperature at
60degC. After cooling to 20 °C, the purple colored product was filtered out over two layers of coffee filter paper, washed once with 50cm3 distilled
water. The moist caked solid was then partially dissolved in >98% alcohol (50mL ethanol was used), then the alcohol was allowed to evaporate out on
an electric hot plate set to only 60 °C. The evaporation should be carried out in the dark, but with plenty of ventilation. About 5 hours are
required for complete evaporation. From this procedure, about 11 grams of nickel hydrazinium nitrate is obtained, which is a 90% yield. Heating of the
reactants/reaction is not in any way necessary, as similar yields were obtained at room temperature, but the product obtained from heating shows
better physical properties, as the salt is of a more crystalline form. The crystalline form has a density of about 0.89 g/cm3. The nickel hydrazinium
nitrate thus obtained, when gradually heated, explodes at 219 °C. The compound appears thermally stable even up to 200 °C. Sensitivity (50%
probability of explosion using 2kg drop hammer from variable heights) value of 84cm. NHN is resistant to friction up to 10N, resistant against
electrostatic discharge, but is sensitive to flame and will explode in contact with a red hot wire. Velocity of detonation: about 7km/s
The co-crystallization of NHN with silver azide, such that the resulting clathrate contains 10% by weight of AgN3, increases the drop height
sensitivity to a value of 66cm. Even such a clathrate containing only 2% silver azide is not much less sensitive, having a drop height value of 68cm.
Cobalt hydrazinium nitrate, which can be similarly prepared, is even more sensitive, having a sensitivity drop height value of 59cm. The cobalt salt
also explodes at a lower temperature, 188 °C. The cobalt salt is, however, actually somewhat less sensitive to friction than NHN.
|
Nitromethane also can form explosive salts with bases:
| Quote: |
When nitromethane is mixed with a concentrated solution of ammonium hydroxide, after several hours the liquid develops a dark brown color and a
crystal substance begins to form. The small crystals formed are colored and difficult to purify.
In a bottle, 20 cc (cm3) of pure nitromethane is mixed with 8 cc of the ammonia solution, and ammonia gas is then bubbled into the mixture
until it is complely saturated (until no more gas can dissolve). A cap is then placed on the bottle and the bottle is kept under 10 °C for about a
day. The crystals which separate out are then removed and gently crushed into a moist powder. The powder is placed on an unglazed clay tile to draw
out the water and allowed to air dry. The residual liquid still in the bottle is again saturated with ammonia gas and the process is repeated as
before. This method will yield about 12 grams of the crystal substance.
The crystals are soluble in methanol, less soluble in ethanol, and nearly insoluble in ether or chloroform. When the crystals are heated, they
decompose, producing some poisonous hydrogen cyanide gas.
The crystals are ammonium 2-nitroethanaloximate, with the formula
NH4+ C2H3N2O3-.
The structure can be written HON=CHCH=NO2(-).
With alcoholic solutions of NaOH, nitromethane forms the simple salt, but upon addition of water, the dark brown red coloration immediately appears,
and the nitromethane is irreversibly resolved to 2-nitroethanaloximate.
|
[Edited on 10-7-2013 by AndersHoveland]
|
|
|
Ral123
National Hazard
Posts: 735
Registered: 31-12-2011
Member Is Offline
Mood: No Mood
|
|
Everyone, permanganate flash, various NPED's, fulminate, picrates, NC at good conditions I've heard of a guy who can initiate stuff with only methyl
nitrate. I may have accidently set off EGDN/NC with modified BP.
|
|
|
Microtek
National Hazard
Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
The key to effective primaries is nitrite. If you can order some, do that. Otherwise, there is a number of ways that you can prepare it at home. It is
a chance to experiment a bit with the technical side of chemistry, but it is quite cumbersome (at least the methods I have tried).
Once you have the nitrite, the rest is relatively easy. Hydrazine synthesis is not difficult, but use the route from urea instead of ammonia (Rosco's
method is excellent).
Apart from azides, there is also nitrotetrazoles, DDNP and nitrobenzenediazonium perchlorates. These are just a handful of good ones that all require
nitrite.
|
|
|
Bot0nist
International Hazard
Posts: 1559
Registered: 15-2-2011
Location: Right behind you.
Member Is Offline
Mood: Streching my cotyledons.
|
|
I am
murcury fulminate
Slilver acetylide
Lead azide
Basic lead picrate
SA\DS is much better than HDMT. Where did you read this?
Remember, all primaries are inherently dangerous. Still, id stay away from peroxides if i were you.
IIRC, MHN has some imtresting properties, and has been employed in DDT detonators before.
[Edited on 10-7-2013 by Bot0nist]
U.T.F.S.E. and learn the joys of autodidacticism!
Don't judge each day only by the harvest you reap, but also by the seeds you sow.
|
|
|
Ral123
National Hazard
Posts: 735
Registered: 31-12-2011
Member Is Offline
Mood: No Mood
|
|
IIRC azide>DS>styphnate>picrate>base picrate>BP>sand?
Does the picrate burn like that one: http://www.youtube.com/watch?v=-ZfwKolJ_Ic
What confinement and amount it requires to initiate, let's say picric acid?
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
Another old primary is "Tetrazene"
| Quote: |
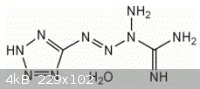
Tetrazene was first prepared by Hoffmann and Roth, but its true structure was only recognized later (Patinkin, Chem. Zentr. 1955. p8377) to be
1-(5-tetrazolyl)-3-guanyl tetrazene hydrate, or alternatively tetrazolyl guanyltetrazene hydrate,
(HN4C)-N=N-N(-NH2)-C(=NH)-NH2*H2O
Lead block test: 155cm3 /10g. Detonation Velocity: around 1500 to over 4000 m/s (depending on how it is initiated). Density: 1.7 g/cm3.
It is a colorless pale yellow, fluffy material with slight hygroscopic properties. It is almost insoluble in water, alcohol, ether, and benzene. It is
stable at normal temperatures when wet or dry, but decomposes in boiling water. Tetrazene is sensitive to friction, shock, and flame. Its brissance is
maximized when it has not been compacted, and this compound can easily become dead-pressed. Tetrazene is not suited for blasting caps or alone as an
explosive since it does not detonate itself very efficiently. It is best suited for booster charges or in blasting caps mixed with other explosives.
It should be detonated by another explosive charge, otherwise if just ignited, it will undergo a lower velocity detonation.
Preparation:
Prepare a solution of 34 g of aminoguanidine bicarbonate and 12.5 mL of glacial acetic acid with 2500 mL of water in a 3-liter Florence flask. Gently
warm the flask on a steam bath and shake periodically until everything is completely dissolved into solution. The solution should be filtered to
remove any impurities that may have not dissolved, then cooled to 30º C by running cold water from the faucet over the flask. It is necessary to
filter the solution if there are impurities present. Add 27.6 g of sodium nitrite to the solution while swirling to dissolve it. Set the flask aside
at room temperature for 3 or 4 hours then shake it vigorously to start precipitation of the product. Let the flask stand for another 20 hours. After
standing, decant as much of the solution off as possible and drown the remaining crystals with water. Decant and drown with water several more times
to wash the crystals. Filter the washed crystals to collect them and thoroughly wash again with water. Dry the product at room temperature and store
in a sealed glass container to keep out the moisture.
Reaction with a strong base causes the compound to hydrolyze into "triazonitrosoaminoguanidine", which is significantly more sensitive. (I
suspect the actual structure is (N3)2C=NH*H2O ). In any case, this ambiguous derivative can be reacted with copper acetate to form a copper
coordination salt, and then with HCl solution (without nitrite this time) to form 5-azido tetrazole in 85% yield.
(1-(5-tetrazolyl)-4-guanyl tetrazene hydrate) is slightly more impact-sensitive than mercury fulminate. When pressed enough, its sensitivity is
reduced or destroyed; this is known as dead pressing. It also decomposes in boiling water. In contact with fire, it readily explodes, producing large
amounts of black smoke. It is prepared by reacting sodium nitrite with an aminoguanidine salt dissolved in acetic acid at 30–40 °C.
(HN4C)N=N--N(NH2)--C(NH)NH2
Nitrate and perchlorate salts of this are probably possible and more powerful.
Treatment of aminoguanidine bicarbonate with sodium nitrite and excess HCl solution makes guanylazide, whereas treatment with a solution of acetic
acid and sodium nitrite forms mostly (HN4C)-N=N-NH-(CN4H), where the (CNH4) is a tetrazole ring.
Guanylazide reacts with sodium hydroxide to form sodium azide, but reacts with a weak base, or weak acid, to form 5-Amino-tetrazole. Nitrous acid
(HONO) oxidizes aminoguanidine nitrate H2NC(=NH)NH2 into guanyl azide N=N=NC(=NH)NH2, which cyclizes into aminotetrazole {HN4C}NH2 when boiled under
alkaline conditions. This takes several hours and gives 70-85% yield.
|
For preparing the Aminoguanidine which is needed to make Tetrazene, there are either two methods. One is to react concentrated hydrazine hydrate with
guanidine, resulting in an ammonolysis reaction. The other is reduction of nitroguanidine:
| Quote: |
Synthesis: Two hundred and sixteen grams (2.07 moles) of nitroguanidine and 740 g. (11.3 moles) of purified zinc dust are thoroughly ground together
in a mortar, and then enough water (about 400 mL.) is added with stirring with the pestle to form a thick paste. The paste is transferred to a 3-l.
enameled can or beaker surrounded by an ice bath. A solution of 128 g. (2.14 moles) of glacial acetic acid in 130 ml. of water is cooled to 5° in
another 3-l. beaker, which is fitted with a strong mechanical stirrer and surrounded by an ice bath. The paste of nitroguanidine and zinc dust, cooled
to 5°, is added slowly with mechanical stirring, the temperature of the reaction mixture being kept between 5° and 15°. A total of about 1 kg. of
cracked ice is added to the mixture from time to time as the mixture becomes too warm or too thick to stir. The addition
of the paste is done over the time of about 6 hours, and the final volume of the mixture is about 1.5 L.
The mixture is then slowly warmed to 40degC on a water bath with continued stirring, and this temperature is maintained for 2–5 minutes, until
reduction is complete. The solution is immediately separated from the insoluble material by filtration on a large Büchner funnel, and the cake is
sucked as dry as possible. The residue is transferred to the 3-L. beaker, washed well with 1 L of water, and then separated from the liquid by
filtration. In the same manner, the residue is washed twice more with two 600-ml. portions of water. The filtrates are combined and placed in a 5-l.
round-bottomed flask. Two hundred grams of ammonium chloride is added, and the solution stirred until they fully dissolve. The ammonium chloride
prevents the coprecipitation of zinc salts when sodium bicarbonate is added to the solution to precipitate the aminoguanidine as the bicarbonate. If
the solution is not clear at this step, it should be filtered.
The stirring is continued, and 220 g. (2.62 moles) of sodium bicarbonate is added during a period of about 10 minutes. The aminoguanidine bicarbonate
begins to precipitate after a few minutes, and the solution is then placed in a refrigerator overnight. The precipitate is collected by filtration on
a Büchner funnel. The cake is removed to a 1-l. beaker and mixed with a 400-ml. portion of a 5% solution of ammonium chloride and filtered. It is
again washed with two 400-ml. portions of distilled water, the wash solution being removed each time by filtration. Finally the solid is pressed down
on the Büchner funnel; the mat is broken up with a spatula and washed while on the funnel with two 400-mL portions of 95% ethanol and then with one
400-ml. portion of ether. After air drying, the aminoguanidine bicarbonate amounts to 180 g. (64% yield) of white solid, melting at 172° with
decomposition. It should not be recrystallized from hot water, since decomposition will occur. The aminoguanidine bicarbonate thus pepared is pure
enough for most purposes.
|
| Quote: | Diazotizing Aminoguanidine
If aminoguanidine is treated with a mineral acid and sodium nitrite, the product is guanyl azide.
NH2C(=NH)NHNH2 + HNO2 --> NH2C(=NH)N3 + (2)H2O
If diazotization is carried out in the presence of acetic acid and sodium acetate, then diazoaminotetrazolic acid results. (HN4C)NHN=N(CN4H) , (where
the structure of the compound contains two tetrazole rings)
In neutral aqueous solutions, tetrazolyl guanyltetrazene hydrate is formed. |
Another interesting reference:
| Quote: | Nitroguanidine reacts with one equivalent of hydrazine hydrate to form nitroaminoguanidine.
R. Henry, R. Makosky, G. Smith, Journal American Chem. Society, 1951, Issue #73, p474. |
[Edited on 11-7-2013 by AndersHoveland]
|
|
|
The_Davster
A pnictogen
Posts: 2861
Registered: 18-11-2003
Member Is Offline
Mood: .
|
|
If you have aminogunanidine and nitrite, do not waste time on a shitty primary like tetrazene. That stuff needs like a gram to initiate tetryl.
Make aminotetrazole, and from there do disilveraminotetrazole perchlorate, or any of a number of nitrotetrazolates.
Silver nitrotetrazolate is what I used to roll with.
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by The_Davster  | | If you have aminogunanidine and nitrite, do not waste time on a shitty primary like tetrazene. That stuff needs like a gram to initiate tetryl.
Make aminotetrazole |
My point was, if you react aminoguanidine with nitrite, you could end up getting either aminoguanidine or Tetrazene if you do not know what
you are doing.
Lead chlorite, Pb(ClO2)2, is another primary. not the best, but easy to prepare
Nitrated sugar alcohols can also be used as primaries. Despite being hazardously sensitive to impact/friction, they are not very reliably initiated by
flame (supposedly nitroglycerin tends to just slowly burn with a pale flame if ignited by a match stick). Still, if the fuse first ignited a little
chlorate pyrotechnic composition, this could then be enough to detonate some solid xylitol pentanitrate, for example.
[Edited on 10-7-2013 by AndersHoveland]
|
|
|
Simbani
Hazard to Self
Posts: 50
Registered: 12-12-2012
Member Is Offline
Mood: No Mood
|
|
I´ve done quiet a bit of energetics in the past, I´m trying to recall the non-peroxide primarys I did.
-silver acetylide, both the pure stuff and various doublesalts
-lead/silver azide
-mercury azide (that is a bitch!)
-silver/mercuryfulminate
-NHN/NHP (perchlorate)
-DDNP (only in minute quantities, no practical usage)
-lead picrate
-copper-ammonia complexes (chlorate/perchlorate, nitrate and persulphate)
If you ask me, go for SA DS and kickstart another primary like NHN or DDNP with it.
My next jouney will be about 5-ATZ,nitrotetrazoles and finally I will try myself on TATNB (I want to make this stuff since 2009). I think this will be
the last energetic compound I will synthesize, I´ve discovered new interests.
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
There is also copper acetylide, though I am not sure whether it would be an effective primary.
| Quote: | Copper(I) Acetylide, Cu2C2
Copper Acetylide is prepared from cuprous chloride, Cu(I)Cl, in Ammonium Hydroxide, by bubbling in acetylene gas.
The copprous salt may be difficult to obtain. One route is to heat CuCl2 over a flame, driving off Cl2 and leaving being CuCl. The CuCl is almost
completely insoluble in water, but in the presence of another chloride salt will dissolve (this can be used as a test to determine if all the chlorine
has been driven out).
Another route is to dissolve copper metal with aqueous chlorine, making sure that the copper is not allowed to completely dissolve, and letting the
reaction sit to be sure that any CuCl2 fully reacts with the remaining copper. (it is actually CuCl2‒ ion that is forming
and dissolving in the solution, the chemistry of copper is much affected by the presence of ligands such as the chloride ion)
Or Cu2O with a little hydrochloric acid.
Copper(I) acetylide is an unusual explosive in that its instant decomposition is self sustained, but does not generate enough heat to vaporize any of
the resultant products, and so there is only traces of gas (from impurities in the compound) to expand besides from the air. It is possible to make
pure Cu2C2 that does not (or only weakly) explodes when initiated under a vacuum. |
I think maybe if just a little bit of potassium chlorate was mixed in as well (10-15%), it could greatly help Cu2C2 to be able to initiate other
explosives. Before the widespread use of lead azide detonators, the detonating caps that used mercury fulminate often had some KClO3 mixed in for this
very reason. I think it may have had to do with better oxygen balance and increasing the temperature. In small amounts and under the confined
conditions in the cap, more energy released may have helped increase the force of the detonation (even though the brisance would be less).
[Edited on 11-7-2013 by AndersHoveland]
|
|
|
golfpro
Banned
Posts: 179
Registered: 18-5-2013
Member Is Offline
Mood: Cap Sensitive
|
|
From the looks of things, SA DS would be easiest, but not the cheapest, somewhere I saw 5g AgNO3 yeilds 3.12g SA. And I can buy 50g of Silver Nitrate
for 80$. This would probably be my only option because I don't have access to nitric acid.. I also do not want to order any, too expensive/risky, I
know I could make it from distillation, but there is 200$ on distilling appuratus.
The one source of Silver Nitrate online for a decent price in 5g amounts won't sell to individuals.
It's all a matter of how much you want to spend and how accesible chemicals are based on where you live.
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
Is this just complete ignorance of chemistry? What is the problem if you can't find AgNO3 ? Just where do you think AgNO3 comes from?
art supply stores often sell silver foil or silver wire, if you cannot find silver anywhere else
Silver is fairly expensive though. At my university, whenever we used AgNO3 in reactions we had to dump it in a big special container afterwards, the
teacher said it would get sent to a recycling company and the school got money for it.
What about just dissolving silver with a mix of 30% sulfuric acid and sodium nitrate? There is no reason the impurities have to separated out, because
the Ag2C2 will still be able to precipitate out when the acetylene is bubbled through (assuming the pH has been neutralized).
The only problem would be when neutralizing you should be careful to only add just enough to neutralize the remaining acid and no more, otherwise you
would start getting AgOH or Ag2CO3 to start precipitating out.
Honestly, I am not sure whether excess acid or the presence of ammonia would prevent the Ag2C2 from being able to precipitate out.
[Edited on 11-7-2013 by AndersHoveland]
|
|
|
golfpro
Banned
Posts: 179
Registered: 18-5-2013
Member Is Offline
Mood: Cap Sensitive
|
|
No, I have silver. I do not have Nitric Acid (or HNO3) to form the Silver Nitrate. I knew silver was expensive. What I am saying is my options for
silver nitrate are down to either having nitric acid to form it from silver or ordering it/buying it. Like I said 5 grams of AgNO3 yeilds just over
3grams of the SA/DS, but good thing only 200-300mg are used per cap w/ a base charge. I'll research that material, and ultimately when I get my CaC2
or acetyline gas and Silver Nitrate I'll make the appuratus for the procedure. I don't care what you say, I would be proud of myself if I got to make
Silver Acetylide Double Salt.
This video has me confused, the guy lights 6 grams of PETN in a plastic container on fire and it explodes? Looks like there is a cap laying on top of
the material though, maybe fire is just for dramatic effect: http://www.youtube.com/watch?v=Lyrj8mKGXEc
[Edited on 10-7-2013 by golfpro]
[Edited on 10-7-2013 by golfpro]
|
|
|
Simbani
Hazard to Self
Posts: 50
Registered: 12-12-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by AndersHoveland |
I think maybe if just a little bit of potassium chlorate was mixed in as well (10-15%), it could greatly help Cu2C2 to be able to initiate other
explosives. Before the widespread use of lead azide detonators, the detonating caps that used mercury fulminate often had some KClO3 mixed in for this
very reason. I think it may have had to do with better oxygen balance and increasing the temperature. In small amounts and under the confined
conditions in the cap, more energy released may have helped increase the force of the detonation (even though the brisance would be less).
[Edited on 10-7-2013 by AndersHoveland] |
IMHO I think KClO3 was used not to enhance the explosive properties, but to fill the voids between the mercury fulminate needles. Pure mercury
fulminate cannot be pressed very good and it "looses" itself up when pure. KClO3 is the best suited for this, inexpensive, non-hygroscopic and
available in great quantitys but it does not hinder the detonation reaction too strong.
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
I still think that adding KClO3 would greatly help the explosive properties of Cu2C2. Let's not forget that a mix of wax and KClO3 can be made to
actually detonate. Combined with copper acetylide, the oxygen released from the chlorate would be able to burn off the carbon, releasing a much
greater amount of energy. Especially since the detonation of Cu2C2 does not even release enough heat to vaporize any of the products, the formation of
gaseous decomposition products would be very helpful.
Copper is also much cheaper than silver, if one wanted to prepare more than very small amounts.
I am not sure about the chemical compatibility, though. It might not be a good idea to store this pre-mixed.
[Edited on 11-7-2013 by AndersHoveland]
|
|
|
caterpillar
Hazard to Others
Posts: 472
Registered: 8-1-2012
Member Is Offline
Mood: No Mood
|
|
I made mercury fulminate (simple preparation), TATP, lead picrate and silver carbide. MF and TATP was successfully used as initiators. I got some shit
instead of Cu2C2 and I read that it is not valuable compound. It you can get any salt of nitric acid, preparation of HNO3 is rather simple. You can
use a teapot for distillation (steel does not react with conc HNO3). Do not use a gum corks -Teflon is good, may be polyvinichloride. Wet asbestos is
good too (and do not tell me shit about its carcinogenic properties). If you need NaNO2, fuse NaNO3 and lead. Obtained PbO fuse with AN, and you'll
get Pb(NO3)2. I suspect, that mixture of MF and KClO3 (plus some other compounds) is used in rifle cups, but I do not think, that aforementioned
mixture is better initiator than pure MF.
Women are more perilous sometimes, than any hi explosive.
|
|
|
golfpro
Banned
Posts: 179
Registered: 18-5-2013
Member Is Offline
Mood: Cap Sensitive
|
|
Cam, it wasn't bubbled through I think it was just the surface exposed to acetyline gas or something I'll have to check.
|
|
|
Ral123
National Hazard
Posts: 735
Registered: 31-12-2011
Member Is Offline
Mood: No Mood
|
|
I think they add KClO3 to MF, to economize the MF...
Just make AP, treat it like nitrogen iodide or something. You'll have reliability and repeatable results. You won't need to purchase stuff. Sometimes
the sellers are stupid enough to ask "what are you going to do with that?". What? I'm giving you my money and you ask stupid questions?
I'm very annoyed at people who don't make basecharges for their initiators, even when they have the stuff. For example 0.3g AP in cartboard, stuffed
in their ETN main.
|
|
|
gnitseretni
Hazard to Others
Posts: 282
Registered: 5-1-2007
Location: Colombia
Member Is Offline
Mood: No Mood
|
|
Note to self: Do NOT take advice from Ral123!
|
|
|
golfpro
Banned
Posts: 179
Registered: 18-5-2013
Member Is Offline
Mood: Cap Sensitive
|
|
Ral's vids are: him lighting a 2 inch fuse while holding his camera and then sprinting 10 meters away, you see a pop, and then he sprints the 10
meters back to instanstly see the results haha
|
|
|
chemcam
Hazard to Others
Posts: 423
Registered: 18-2-2013
Location: Atlantis
Member Is Offline
Mood: I will be gone until mid-september, on a work contract.
|
|
You should be grateful his videos are on YouTube for you to enjoy and learn from. His stuff is practical and what inspired me to make my channel.
|
|
|
scottjm
Harmless
Posts: 17
Registered: 29-6-2013
Member Is Offline
Mood: Recrystallizing
|
|
The best cap I have been able to think of is very easy to make, and is almost primary free. You should be able to make and recrystallize ETN? You will
also need flash. I like SS tube, but Al also works. (you might not like using metals as the case for a cap, but it is very easy to ground unlike
plastic or paper). Seal of one end (I should not have to explain how to do that), with the tube grounded pour in the ETN. Press the ETN until compact.
Fashion and install a small separator disk made out of either Al foil or paper. Pour in your flash powder, and seal up that end. How you seal up the
one end depend on personal preference, and weather or not you are using e-match.
How much flash and how much ETN is dependent on A: what you intend to initiate, and B: How fast you flash is. (Note stick to 7:3 KClO4/Al flash. Other
flashes can be very dangerous) what I use
To initiate a ETN, or PETN booster : 850-1,000 mg ETN/ 1000-2000 mg flash (a bit over kill, but it always get the job done)
To initiate AN/NM/Al : 900-1200 mg ETN/ 1000-2000 mg flash
To initiate chedite : 350-450 mg ETN/ 800-1500 mg flash
You get the idea
|
|
|
Ral123
National Hazard
Posts: 735
Registered: 31-12-2011
Member Is Offline
Mood: No Mood
|
|
If I have to make NPED, it would be something like that.
<!-- bfesser_edit_tag -->[<a href="u2u.php?action=send&username=bfesser">bfesser</a>: removed
unnecessary quote(s)]
[Edited on 7/11/13 by bfesser]
|
|
|
golfpro
Banned
Posts: 179
Registered: 18-5-2013
Member Is Offline
Mood: Cap Sensitive
|
|
Right now I am playing around with things around the house. I am doing the nurdrage experiment making nitric acid (small amount) by bubbling nitrogen
dioxide gas throough 12% H2O2 using 9M HCl approx. and 30 grams of copper. There is a more efficient way to do it still without distillation, but I'll
have to get some more glassware first.
Do you need conc. Nitric Acid to dissolve silver and create the silver nitrate? Or will a slightly weak nitric acid solution disolve silver with heat,
weak nitric acid made by bubbling NO2 can disolve copper, I'll have to try and see.
|
|
|
| Pages:
1
2 |
|









