Bedlasky
International Hazard

Posts: 1251
Registered: 15-4-2019
Location: Period 5, group 6
Member Is Offline
Mood: Volatile
|
|
Simple guide for analytical chemistry
I've decided to make this guide for those of you who are new in analytical chemistry to make your beginnings easier. Analytical chemistry can be a
useful tool for various experiments - you can determine purity of your compound, you can determine contaminants, you can determine reaction products
etc.
Analytical chemistry can be divided into:
Qualitative analytical chemistry - you determine what exactly is in your sample
Quantitative analytical chemistry - you determine how much of some component is present in your sample
For qualitative analytical chemistry you usually don't need some special equipment. However for quantitative analytical chemistry you need some
special glassware/tools. I discuss them below.
We can differentiate between classical methods and instrumental methods. Classical methods are using traditional old
school methods like titrations, gravimetry, test tube tests etc. Instrumental methods are using some more complex machinery for analysis - like
chromatographs, spectrophotometers, mass spectrometers etc. In this topic I will be mostly focused on classical methods, however I have plans to
discuss some basic instrumental methods accessible to amateur chemists (potentiometric and conductometric titrations, refractometry,
spectrophotometry/colorimetry).
Qualitative analytical chemistry is further divided into:
Qualitative inorganic analytical chemistry
Qualitative organic analytical chemistry
Classical qualitative inorganic analytical chemistry mostly uses the hydrogen sulfide system of cationic classes (however there is
also ammonia system) and anionic classes. I will describe the H2S cationic system and anionic classes in separate
thread. These systems however don't contain large amounts of elements (for example most 5th and 6th row transition metals are missing), so I will try
to add tests for some of the missing elements. H2S cationic systems divide cations into 5 classes based on their insoluble salts - I. insoluble
chlorides, II. insoluble sulfides precipitating from acidic solution (this class is then divided into two subclasses based on solubility of sulfides
in ammonium polysulfide), III. insoluble sulfides precipitating from alkaline solution, IV. insoluble carbonates, V. other cations. Anionic system
divides anions according to reactions with these reagents: precipitation with Ag+ and Ba2+, redox reactions with MnO4-, I2 and I- and reactions with
non-oxidizing acids. Again, lots of anions are missing in this system, so I will try to make tests for some other anions.
Classical qualitative organic analytical chemistry is divided into 3 groups:
Elemental analysis - tests for individual elements in organic compound (C, H, O, N, S, halogens)
Functional group analysis - test for individual functional groups in organic compound (-OH, -NO2, -COOH etc.)
Measurement of physical constants - measuring of melting/boiling point, density, viscosity, refractive index and specific rotation,
calculation of refractive index
Classical quantitative analytical chemistry is further divided into:
Gravimetry - determination of the component using precipitation and weighing precipitate of exact composition
Volumetry - determination of the component using titration with solution of exact concentration
Volumetry is further divided into:
Acido-basic titrations titrations using acid or base as titrants. Further divided into acidimetry (acid as titrant)
and alkalimetry (base as titrant)
Redox titrations titrations using oxidizing or reducing agents as titrants. Further divided into oxidimetry
(oxidizing agent as titrant) and reductometry (reducing agent as titrant)
Complexometric titrations titrations using complexing agent as titrant
Precipitation titrations titrations using as titrant some compound which forms insoluble compound with the component in the sample
These groups are further divided mostly into these methods (plus some exotic methods):
Acidimetry:
Titrations with HCl, H2SO4
Alkalimetry:
Titrations with NaOH, KOH
Oxidimetry:
Manganometry - KMnO4 as titrant
Dichromatometry - K2Cr2O7 as titrant
Bromatometry - KBrO3 as titrant
Cerimetry - Ce(SO4)2.4H2O or (NH4)4Ce(SO4)4.2H2O as titrant
Iodometry - using I2 in the solution of KI as titrant
Reductometry:
Ferrometry - using FeSO4.7H2O or (NH4)2Fe(SO4)2.6H2O as titrant
Titanometry - using TiCl3 as titrant
Chromometry - using CrCl2 as titrant
Iodometry - using back titrations with Na2S2O3.5H2O as titrant + KI as I2 generating reagent
Complexometry:
Chelatometry - using Na2H2edta.2H2O as titrant
Mercurimetry - using Hg(NO3)2 as titrant
Precipitation titrations:
Argentometry - using AgNO3 as titrant
For these methods you need some special equipment. For both gravimetry and volumetry you need decent scales - this is your most important equipment
for analysis. Ideally analytical scales with 4 decimal places. However these scales are quite expensive and for amateur chemistry purposes you will do
just fine with some less accurate scales. Ordinary scales with 2 decimal places are absolute minimum for decent
analysis, however I think that scales with 3 decimal places are more optimal. The more accurate scales you can get,
the better. More accurate scales = smaller error in the determination. It all depends on your financial situation and for what purpose you need
analysis.
Volumetry and gravimetry then need different special equipment. Gravimetry needs far less equipment, however it is more tedious and you can more
easily introduce some error during precipitation. Volumetry is faster and easier to handle, but needs more equipment. I list all special things you
need in an ideal lab, but lots of stuff can be replaced with something else - it all depends on your creativity. But if you are replacing something,
be sure it doesn't ruin your analysis. I don't list some common equipment like beakers, stirring rods etc. Stuff that is very important is highlighted
Equipment for gravimetry:
Analytical funnel - funnel with spiral grooves. It makes filtration quicker due to faster removal of the filtrate through grooves. It
is made from glass or plastic.
Ashless filter papers with a given size of pores - these are special filter papers for quantitative analysis. Sometimes you need to
thermally decompose your precipitate into something else, so you burn your sample with filter paper - this would introduce some mistake because of ash
from filter paper. These filter papers have minimal amount of ash after burning (0,05-0,13 mg in the circle with 9-12,5 cm diameter). In Europe they
are sold with three pore sizes - 388 with red overprint (3,7 µm pores), 389 with yellow or white overprint (3,4 µm pores) and 390 with blue
overprint (1,1 µm pores). Different pores are suitable for different precipitates. Most widely used are probably the blue ones. I
personally never worked with red filter papers.

Glass filter crucible - also with different pore sizes, mostly widely used are S3 (30 µm pores) and S4 (8 µm pores) crucibles
Porcelain crucible - for burning precipitates
Some kind of burner (Bunsen, alcohol etc.)
Some determinations use burette, but this can be replaced with pipette or separatory funnel (you don't need to add exact amount of reagent)
Equipment for volumetry:
Burette - 25 or 50 ml is best for most purposes
Volumetric flask - 50 and 100 ml are most universal, 0,5 or 1 l are useful for making stock solutions. You need at least 2-3 small
flasks.
Glass pipette - ideally you need bulb pipettes, 5, 10, 25 ml are most universal. You can use graduated pipettes instead, but they are
less accurate. There are also automatic pipettes, but these are far more expensive than the glass ones.
Titration flask - wide neck flat bottom flask, wide neck gives you enough space for comfortable titration. You can use some other flask, but a thin
neck gives you less space for mixing and titrating at the same time. You can also use a beaker, but there is a higher chance of splashing solution
outside, which causes error in determination.
Erlenmayer flask with ground glass joint - these are useful in iodometric and bromatometric back titrations, preventing I2 and Br2 loss during
reaction time.
Weighing pan - useful for weighing and transferring reagents, but they are not necessary
Hopefully you find this summary useful. Next time I will describe some basic procedures in gravimetry and volumetry. If you want to cover some topic
in which you are interested, let me know.
[Edited on 14-1-2024 by Bedlasky]
[Edited on 3-6-2024 by Texium]
|
|
|
Admagistr
Hazard to Others
Posts: 383
Registered: 4-11-2021
Location: Central Europe
Member Is Offline
Mood: The dreaming alchemist
|
|
Hi that'super, great project! I would be interested in evidence of metal cations! Especially the lesser known and rarer elements like beryllium,
gallium, hafnium, lanthanides...
|
|
|
Bedlasky
International Hazard
Posts: 1251
Registered: 15-4-2019
Location: Period 5, group 6
Member Is Offline
Mood: Volatile
|
|
I haven't experiment with much of these rarer elements. I have done quite extensive research on molybdneum and tungsten:
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
I have some plans doing Nb, Ta and Zr chemistry, I found some interesting colorful tests for Nb, Ta, Zr, Hf and Ti (if everything works fine for Zr, I
buy some Hf and try that with Hf). I also have some plans for Re and Ru chemistry.
[Edited on 14-1-2024 by Bedlasky]
|
|
|
Admagistr
Hazard to Others
Posts: 383
Registered: 4-11-2021
Location: Central Europe
Member Is Offline
Mood: The dreaming alchemist
|
|
So I'll look forward to seeing the metals you have experience with and have a plan for
|
|
|
Texium
|
Thread Topped
14-1-2024 at 16:09 |
Bedlasky
International Hazard
Posts: 1251
Registered: 15-4-2019
Location: Period 5, group 6
Member Is Offline
Mood: Volatile
|
|
Gravimetry
Gravimetry is based on precipitation of some ion we want to determine from its aqueous solution and weighing that precipitate. Precipitate is then
filtered, washed and dried or ignited. Precipitate must have precise, well defined chemical composition. Example of such precipitate is BaSO4. For
example Fe(OH)3 doesn't have precise, well defined composition and must be ignited to dehydrate it into Fe2O3 which has exact composition. This final
form (BaSO4, Fe2O3...), which is then weighed in order to determine content of some ion, is called the weighing form.
Precipitation isn't as easy as it looks at first sight. We must consider several aspects which affect solubility and purity of the precipitate and the
filtration process. We want from the precipitate to have sufficiently low solubility, exclude as much impurities as possible and to form as big
particles as possible.There are three kinds of impurities that can form during precipitation: inclusion, occlusion
and adsorption.
Inclusion is when another ion with similar size and charge replaces a determined ion in the crystal lattice of precipitate. Inclusion is chemically
part of the precipitate, so it can only be removed by reprecipitation. Precipitating from dilute solution decreases the chance of
inclusion, because there are less ions per volume of the solution. During precipitation of hydroxides there is also a chance that alkali metal ions
are part of the structure (for example in MnO2 or Fe(OH)3) - this is the reason why NaOH or KOH aren't used as precipitants. Ammonia, which can be
easily removed, is used instead.
Occlusion is when impurity is physically trapped inside the precipitate. Occlusion can be minimized by digestion - heating the
precipitate in the supernatant solution for prolonged time. Precipitate constantly dissolves and reforms, so impurity is eventually exposed to the
solution and removed from the precipitate. Precipitate grows again slowly during this process, so there is a smaller chance for formation of another
occlusion. Some precipitates also change their structure from amorphous to crystalline during digestion. Precipitating from dilute solution also
minimizes the chance of occlusion. Constant stirring is also another important factor - this prevents local high concentrations of impurities.
Adsorption is when impurity sticks to the surface of precipitate due to intermolecular forces. We can minimize adsorption by growing bigger particles
of precipitate. This can be achieved by digestion, because reforming particles are created slowly and grow bigger. Also the slower initial
precipitation is, the bigger particles get. Slow rate of reaction can be achieved by adding substance that can be slowly converted into
precipitant. For example hydroxide can be generated by hydrolysis of urea, sulfide can be generated by hydrolysis of thioacetamide or
thiourea, chromate can be generated from Cr3+ and bromate, sulfate can be generated by hydrolysis of sulfamic acid etc. Another factor is
coagulation of electrically neutral particles. For example if we precipitate Ag+ as AgCl by adding NaCl to the AgNO3 solution, then
AgCl particles adsorb on their surface primary layer of Ag+ ions. This first layer attracts NO3- ions which form the secondary layer. This thick
secondary layer prevents AgCl particles from coming together. When we keep adding NaCl, more Ag+ ions are converted into AgCl which decreases the
amount of adsorbed Ag+, which in return decreases the thickness of the secondary layer. But the solution isn't to add as much NaCl as possible,
because when excess NaCl is added, Cl- ions are adsorbed onto AgCl particles and Na+ ions create a secondary layer. So it's important not to add a
huge excess of precipitant. Another thing which helps with coagulation of particles is precipitation from hot solution, because increasing temperature
causes decrease of adsorbed ions.
So from these informations we can summarize what is important for correct precipitation:
1. Precipitate from dilute solution (by dilute solution of precipitant)
2. If large amount of ions with similar size and charge as our analyte (determined substance) are present (for example Co2+ and
Ni2+), we should consider reprecipitation (or masking interfering ion)
3. Precipitate from constantly mixing solution
4. Digestion of precipitate for prolonged time (usually around half an hour)
5. Precipitating from hot solutions (unless it is undesirable)
6. Using precipitants with volatile ions, if it is possible (ammonium salts, nitrates etc.)
Another step is filtration. You need filter paper or filtration crucible with the right size of pores (this is discussed in the post above).
Filtration is usually performed when the suspension is still hot, because filtration is much quicker. Firstly you decant as much liquid as possible
(through filter paper of course). Then you filter the last portion containing your precipitate and wash and transfer the rest of the precipitate onto
the filter paper with water. Do it slowly using a glass stirring rod to avoid splashing. This picture describes it better than a thousand words:
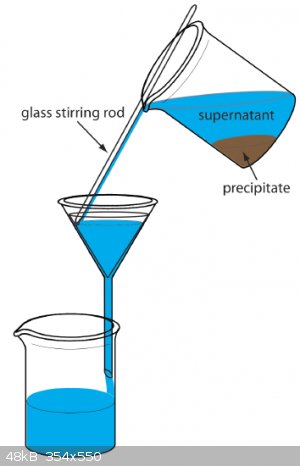
Then we rinse the precipitate with several small portions of solvent (usually water) to remove all soluble ions. It's helpful if you test filtrate for
these ions - for example chlorides using AgNO3 or sulfates using BaCl2. Some precipitates are prone to peptization - converting big
particles to smaller particles - during washing with water (this is usually the case of hydroxides). These precipitates are washed with inert volatile
electrolyte which is later removed during ignition (for example Fe(OH)3 is washed with NH4NO3 solution). Precipitates containing acidic or basic ions
may be sensitive to changes in pH, so it's also important to consider this factor.
Final step of gravimetric analysis is converting precipitate to its weighing form - that means drying or igniting the precipitate. Which one of these
two you choose depends on if your precipitate precipitates out of solution with precise chemical composition or not. Precipitates like AgCl, BaSO4,
BaCrO4, Ni-diacetyldioximate etc. have precise composition, so you can just dry them (together with filter paper). In this case, before you even start
precipitating, you need to dry filter paper at 105 °C, let it cool in a desiccator and then immediately weigh it as fast as possible. Filter paper
absorbs moisture from the air, so the longer it stays on the air, the bigger error in your determination will be. After drying filter paper with
precipitate at a certain temperature (usually within the range 100-130 °C), you let it cool in a desiccator and then weigh it as quickly as possible.
By subtracting the weight of the filter paper from the weight of filter paper with precipitate you get the weight of the precipitate.
Precipitates like CaSO4.xH2O, SiO2.xH2O, Fe(OH)3, SnS2 etc. don't have exact chemical composition and needs to be ignited in order to convert them to
compounds with exact chemical composition (CaSO4, SiO2, Fe2O3, SnO2 etc.). Advantage of this method is that you don't need to weigh dry filter paper,
because it will be burned during heating. You need to weigh the crucible beforehand. Crucible must be firstly dried at 105 °C and then cooled in a
desiccator, because porcelain still adsorbs a little bit of moisture (not much, but still...).
Some precipitates need special methods - for example HgS contain some elemental sulfur which must be extracted with toluene before drying the
precipitate. CdS doesn't have exact chemical composition but cannot be simply ignited to form CdO, because CdO also doesn't have exact chemical
composition. So it is instead boiled with concentrated H2SO4 to the dryness and converted into CdSO4.
Determination of calcium is an interesting one, because you precipitate it as CaC2O4.H2O. You can then dry it at 105 °C or heat it at 500 °C or 900
°C to form CaCO3 or CaO. All three compounds are weighing forms.
Lastly, I would like to discuss calculations. Gravimetric calculations are usually easy and straightforward. I give an example:
How much cadmium is in a sample of an alloy? Cadmium was precipitated as NH4CdPO4.H2O, ignited and weighed as Cd2P2O7. Weight of Cd2P2O7 is 0,3204 g,
weight of the alloy is 1,7303 g.
Firstly we need to find how much Cd is in Cd2P2O7. Mass fraction of Cd can be simply calculated from molar masses:
w(Cd)=2*M(Cd)/M(Cd2P2O7)
It's important not to forget that Cd2P2O7 contains 2 Cd atoms during this calculation. So from this we can calculate mass of Cd:
m(Cd) = m(Cd2P2O7)*2*M(Cd)/M(Cd2P2O7)
Now we know the mass of Cd. So what is the mass fraction of Cd in alloy?
w(Cd) = m(Cd)/m(alloy) = m(Cd2P2O7)*2*M(Cd)/[M(Cd2P2O7)*m(alloy)]
w(Cd) = 0,3204*2*112,414/(398,769*1,7303) = 0,1044 = 10,44 %
We can generalize formula for calculations (AxBy is weighing form):
w(A) = m(AxBy)/m(sample) * x*M(A)/M(AxBy)
Part x*M(A)/M(AxBy) is also called gravimetric factor and is denoted by fg. So formula in short form is:
w(A) = m(AxBy)/m(sample) * fg
And finally here is spreadsheet of some basic determinations:
Attachment: Gravimetric determinations.xlsx (12kB)
This file has been downloaded 197 times
Sources:
https://web.vscht.cz/~koplikr/Část%20A3.pdf
https://chem.libretexts.org/Bookshelves/Analytical_Chemistry...
https://www.researchgate.net/publication/278032531_Thermal_b...
[Edited on 3-6-2024 by Texium]
|
|
|
teodor
International Hazard
Posts: 1002
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
Thanks Bedlasky for starting this thread. I myself try to study inorganic analysis and I already have collected several interesting books which
describe methods alternative to the classical H2S. It is still not one system, but may be one day I will find a way for general cation separation. As
usual I am limited with time doing a lot of other activities besides chemistry. But if you are interested we can plan some experiments based on the
methods from those books:

|
|
|
Bedlasky
International Hazard
Posts: 1251
Registered: 15-4-2019
Location: Period 5, group 6
Member Is Offline
Mood: Volatile
|
|
Volumetry
Volumetry is a method based on titration of a sample in the solution. Titration is addition of a solution of reagent with known
concentration (titer) from burette to the solution of a sample in the titration flask. The reagent is called
titrant.
Volumetry is further divided based on the type of reaction between titrant and sample (acido-basic, redox, complex, precipitation) and then based on
what compound titrant is (KMnO4 - manganometry, KBrO3 - bromatometry etc.). More about that in introductory post.
First we start with preparation of stock solution of titrant and determination of its concentration
(standardization). Some titrants need to be standardized because they cannot be made in sufficient purity. This might be due to
hygroscopicity (NaOH, H2SO4), volatility (HCl), photosensitivity (AgNO3, KMnO4), because they are oxidized by oxygen (TiCl3, FeSO4), because they
decompose in the solution (KMnO4), because they react with CO2 (NaOH, KOH) or because they have variable amount of water of crystallization
(Na2H2edta.2H2O), Ce(SO4).4H2O).
For this reason there are primary and secondary standards. Primary standard is chemically pure, non-hygroscopic
substance, which doesn't decompose on light and in the solution, which doesn't react with the components of the air and have precise chemical
composition. Secondary standard doesn't fulfill some of these criteria, but its reaction with titrant is convenient for standardization. A secondary
standard must be standardized with the primary standard. Here is list of the most common titrants with their primary and secondary standards:
NaOH, KOH - oxalic acid dihydrate, sulfamic acid, potassium tetraoxalate dihydrate, potassium hydrogen phthalate (dried at 105 °C), (benzoic acid,
salicylic acid for titrations in alcoholic environment)
HCl, H2SO4 - tris, sodium oxalate, sodium carbonate (anhydrous, dried at 105 °C, sometimes prepared by heating NaHCO3 or Na2C2O4), sodium/potassium
bicarbonate, sodium oxalate (dried at 105 °C)
KMnO4 - oxalic acid dihydrate, sodium oxalate (dried at 105 °C), potassium tetraoxalate dihydrate
Ce(SO4)2, (NH4)4Ce(SO4)4 - oxalic acid dihydrate, sodium oxalate (dried at 105 °C)
I2 - ascorbic acid, As2O3, sodium thiosulfate (secondary standard)
Na2S2O3 - K2Cr2O7, KBrO3, KIO3 (all 3 dried at 105 °C) + KI (liberated iodine is titrated with Na2S2O3)
TiCl3, CrCl2, FeSO4, (NH4)2Fe(SO4)2 - K2Cr2O7 (dried at 105 °C)
Na2H2edta - Pure elemental Cu, Bi, Pb, PbCl2 (dried at 105 °C), CaCO3 (dried at 105 °C), BaCO3 (dried at 105 °C) (carbonates are dissolved in HCl,
from this you make stock solution for standardization)
MgSO4, ZnSO4 - Na2H2edta (secondary standard)
Hg(NO3)2, Hg(ClO4)2 - NaCl, KCl
AgNO3 - NaCl, KCl
KSCN, NH4SCN - AgNO3 (primary or secondary standard depending on the purity)
Some titrants are already sold in sufficient purity to not need a standardization (and they are often used as primary standards as well). These are
K2Cr2O7 and KBrO3. AgNO3 could also be a primary standard, depending on the purity of the product (20 years old AgNO3 isn't a reliable primary
standard). Solution of AgNO3 made from pure silver metal also doesn't need standardization. Iodine also could be a primary standard, if it is
resublimed. If Hg(NO3)2 is prepared from pure elemental mercury, you also don't need standardization. K2Cr2O7, KBrO3 and AgNO3 must be dried at 105
°C before weighing (or it's at least usual procedure, if you don't dry K2Cr2O7, KBrO3 or KIO3, you barely notice difference in concentration).
Stock solutions of titrants are prepared simply by weighing the compound and dissolving it in the water. Resulting solution is quantitatively
transfer into the volumetric flask (you pour the solution into the flask and wash the beaker several times with water) and then the flask is
filled with water to the mark. Compounds which require standardization can be weighed just to 2 decimal places. However primary standards need to be
weighed precisely to 4 decimal places (if you own such accurate scales, see the introductory post).
Now we discuss sample preparation. If you have a soluble solid sample, you just simply weigh it, quantitatively transfer it into the beaker, dissolve
it, transfer into the volumetric flask and fill to the mark with water. Or instead of making a stock solution of your sample in volumetric flask, you
can directly transfer it into the titration flask and dissolve it there. It depends on what's convenient for that particular analysis. If you have a
liquid sample, the procedure is the same, but instead of weighing you use pipette. If you have an insoluble solid sample, you weigh it, transfer it
into the titration flask without washing the weighing pan. You then weigh the pan with residue of the sample. Weight of sample - weight of residue =
weight of sample in the titration flask. This type of weighing is called differential weighing.
Right pipetting is important. Bulb pipettes are more accurate than graduated pipettes. Don't use graduated cylinders for measuring volume, because
cylinders are far less accurate than pipettes. If you look at a pipette, you see these data: Time you should wait until all liquid adhering to the
glass falls down, temperature at which volume was calibrated, volume, deviation from declared volume. These data inform you about the best conditions
for pipetting. When pouring liquid from the pipette, don't push the last drop from the tip of the pipette - volume is calibrated for liquid exiting
the pipette just under force of gravity. If you blow that drop from the pipette, you have greater volume of the sample in the titration flask (by this
you add another error to your determination).
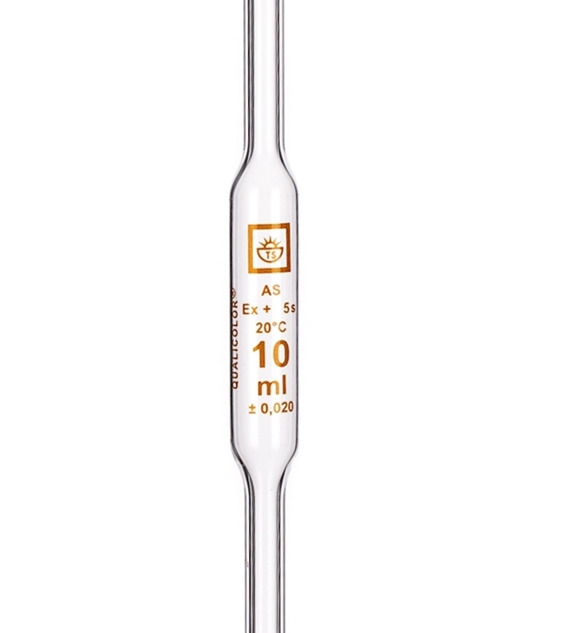
We also need some way to know when the reaction is finished - in other words, how we find a end point. There are two
indications of end point - visual using indicators and instrumental using some device (potentiometers, conductometeres, ammeters,
thermometers etc.). Visual indication is based upon change in colour of the indicator, instrumental indication is based on determination of the end
point from the graph.
Indicators can be divided into six groups:
Acid-base: Methyl orange, methyl red, bromophenol blue, congo red, bromothymol blue, phenolphthalein, thymolphthalein, alkali blue
6B, crystal violet etc.
Redox: reversible - diphenylamine, diphenylamine sulfonate (Na or Ba salt), ferroin, Fe(II)-bipyridyl, Ru(II)-bipyridyl,
methylene blue, indigo carmine, Fe(III)-SCN-, starch-I3-
irreversible - methyl red, methyl orange
Complexometric: metallochromic - eriochrome black T, murexid, xylenol orange, fluorexon, PAN, salicylic acid
others: Diphenylcarbazon, Fe(III)-SCN-
Precipitation: K2CrO4, sodium nitroprusside, KI
Adsorption: Fluorescein, dichlorofluorescein, eosin Y, eosin B, bromophenol blue, congo red, alizarin red S, bromothymol blue
Choosing the right indicator will be described in posts for particular methods.
Potentiometric titration curves use several methods for end point determination. Graphic methods include - method of three parallels, method of three
tangents, method of two circles and Tubb's method. All these methods are in principle the same - you try to find a line which goes through the center
of a titration curve, either by using average distance between two lines or by joining centers of two circles. Again, details will be described
elsewhere. More precise are derivative methods: method of the first derivation and method of the second derivation (which is the most accurate method
of end-point determination).

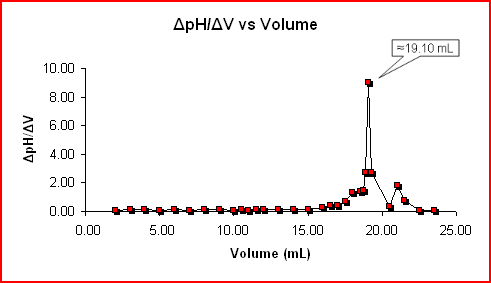
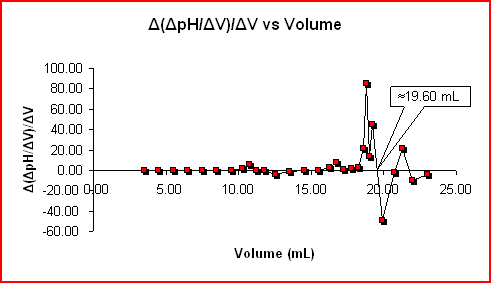
In the conductometric titration curve the linear parts of the curve are elongated and the point where they cross marks the end point.
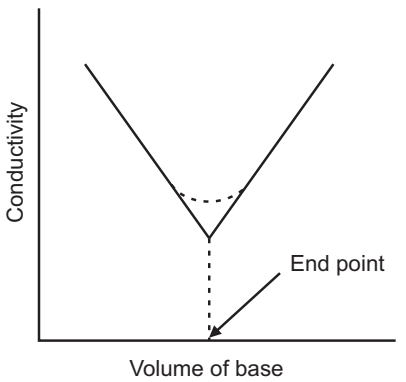
Amperometric titrations have similar curves to potentiometric curves, thermometric titrations have similar curves to conductometric curves, so methods
of end point determination are the same as in potentiometric/conductometric titrations.
Every titration has its own conditions at which the reaction is quantitative (for example titration of Fe2+ with KMnO4 must be performed in a strongly
acidic solution). At the end of the titration it is important to add titrant slowly in order to avoid over titration (adding too much
titrant). Some indicators don't change colour immediately upon reaching the end point, but have slight delay (this is typical for the starch-iodine
complex for example). Near the end point indicators also change colour more slowly. In the case of instrumental indication, measured values sharply
change near the end point (slow down or rise sharply), so it takes longer to reach equilibrium.
Sometimes the sample is too unstable at titration conditions, so it must be added from a burette to the known quantity of titrant. Great example is
manganometric determination of nitrite. Nitrite decomposes in acidic solution to NO and NO2 gases, so it must be added from burette to acidified
permanganate solution to avoid its decomposition.
Sometimes the sample doesn't have a well defined reaction with titrant or there isn't any suitable indicator for direct titration. In
this case we use excess of another compound which reacts with the sample to form some product. This product is then titrated with titrant. This method
is called back titration. These titrations are often used in iodometry. Some oxidizing agents (iodate, hypochlorite etc.) react with
iodide to produce iodine. Iodine is then titrated with Na2S2O3.
Calculations aren't usually difficult. Here are two examples:
1. 2,0435 g of stainless steel was dissolved in sulfuric acid. Solution was transferred into the 50 ml volumetric flask and filled with water to the
mark. 10 ml of solution was transferred into the titration flask, diluted with water and acidified with 10 ml of 1+4 H2SO4. Sample was titrated with
K2Cr2O7, c = 0,1024 mol/l. Two titrations were performed. V1 = 8,3 ml, V2 = 8,5 ml. How much iron is in the sample?
Firstly we must solve equation for this reaction:
Fe2+ + Cr2O72- + H+ --> Fe3+ + Cr3+ + H2O
Fe2+ release 1 electron, Cr2O72- consume 6 electrons. So for every mole of dichromate we need 6 moles of
ferrous ions.
6Fe2+ + Cr2O72- + 14H+ --> 6Fe3+ + 2Cr3+ + 7H2O
From this equation we can make formula for content of iron:
n(Fe)/6 = n(K2Cr2O7)
m(Fe)/(6M(Fe)) = c(K2Cr2O7)*V(K2Cr2O7)
m(Fe) = 6*c(K2Cr2O7)*V(K2Cr2O7)*M(Fe)
w(Fe) = m(Fe)/m(sample) = 6*c(K2Cr2O7)*V(K2Cr2O7)*M(Fe)/m(sample)
Because we only add 1/5 of the sample into the titration flask (10 ml from 50 ml solution), we must multiply the equation by 5.
w(Fe) = 6*c(K2Cr2O7)*V(K2Cr2O7)*M(Fe)*5/m(sample)
Because we perform two titrations, we must make two calculations and take the average of these two results. Or we can make our lives easier and take
the average of volumes (8,4 ml) and have the result in one calculation. Don't forget to write volume in litres, because we calculate with
concentration in mol/l.
w(Fe) = 6*0,1024*0,0084*55,845*8/2,0435 = 0,7052 = 70,52 %
Formula above can be also rewritten as:
w(Fe) = c(K2Cr2O7)*V(K2Cr2O7)*M(Fe)/m(sample) * 6/1 * 50/10
6/1 is molar ratio of Fe2+/Cr2O72-, 50/10 is ratio volume ratio of stock solution/amount pipetted into the
titration flask. Molar ratio is also called titration factor ft, volume ratio is called dilution factor
df.
w(Fe) = c(K2Cr2O7)*V(K2Cr2O7)*M(Fe)/m(sample) * ft*df
I personally don't use these factors in calculations, because it isn't intuitive, you must remember what is in the numerator and what is in the
denominator. But someone might find it easier with these factors.
2. Sample of cerussite was dissolved in acetic acid. Ammonia buffer, 20 ml of 0,0491M EDTA solution and small amount of eriochrome black T were added
into the titration flask. Excess of EDTA was titrated using MgSO4, c = 0,0511 mol/l. Two titrations were performed. V1 = 7,7 ml, V2 = 7,8 m1 = 0,1641
g, m2 = 0,2005 g. How much lead sample of cerussite contain?
Making chemical equation is pretty easy in this one:
Pb2+ + H2edta2- --> [Pb(edta)]2- + 2H+
H2edta2- + Mg2+ --> [Mg(edta)]2- + 2H+
Again, you can calculate for each titration separately, but I make it easier and make just one calculation with average volume (6,5 ml) and average
mass (0,1823 g). First of all, we need to find how amounts of substances of Pb and Mg are connected.
n(EDTA, total) = n(EDTA)1 + n(EDTA2)
n(Mg) = n(EDTA1)
n(Pb) = n(EDTA2) = n(EDTA, total) - n(EDTA1) = n(EDTA, total) - n(Mg)
m(Pb)/M(Pb) = c(EDTA)*V(EDTA) - c(Mg)*V(Mg)
m(Pb) = [c(EDTA)*V(EDTA) - c(Mg)*V(Mg)]*M(Pb)
w(Pb) = m(Pb)/m(sample) = [c(EDTA)*V(EDTA) - c(Mg)*V(Mg)]*M(Pb)/m(sample)
Because samples were weighed and transferred directly into the titration flask, there is no need to consider dilution.
w(Pb) = (0,0491*0,02 - 0,0511*0,0065)*207,2/0,1823 = 0,7386 = 73,86 %
|
|
|
Bedlasky
International Hazard
Posts: 1251
Registered: 15-4-2019
Location: Period 5, group 6
Member Is Offline
Mood: Volatile
|
|
Quote: Originally posted by teodor  | Thanks Bedlasky for starting this thread. I myself try to study inorganic analysis and I already have collected several interesting books which
describe methods alternative to the classical H2S. It is still not one system, but may be one day I will find a way for general cation separation. As
usual I am limited with time doing a lot of other activities besides chemistry. But if you are interested we can plan some experiments based on the
methods from those books:
|
That would be great Teodor! I was also considering making such a system. It is also possible that a simple system for "all" cations cannot be made.
Chemists failed to make such a system for anions. Another thing is that there are plenty of complex cations like [Co(NH3)6]3+, [Co(NH3)5Cl]2+ etc. But
I would probably stick only with "simple" cations, complex cations would make it very difficult. I plan as my next post identification of a mixture of
cations using the H2S system. I never did it before, I only tried individual cations at school.
|
|
|
RU_KLO
Hazard to Others
Posts: 245
Registered: 12-10-2022
Location: Argentina
Member Is Offline
|
|
Im too in analytical chemistry, and am trying to device a non sulfide system, which is a frankenstein of other schemes.
But as I dont have to much knowledge (only DIY) no precise stantandards and equipment, its a bit difficult for me.
This is "my" proposed Scheme:
Group/ cations / Agent / Notes
0 / Na+; K+; Nh4+/ none / single test
1 / Hg+, Ag+, Pb++, Tl+, WO4--/ Hcl / Chloride Group - from most schemes
2 / Fe+++; Bi; Cr+++; Al; Sb; Sn / NH4Bz + NaBz / Basic Benzoates group
3 / Pb, Ba; Sr; Ca / H2SO$ / sulfate group
4 / Mn, Fe, Hg, Cu; Ni, Cd, Co / NaOH / precipitate nonamphoteric group
5 / Sn; As, Zn / NaOH / solution amphoteric group
Mg is missing, but in most schemes it is precipitated at the end as phosfate.
Go SAFE, because stupidity and bad Luck exist.
|
|
|
j_sum1
Administrator
Posts: 6374
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
There is a video out there "testing for cations" or some similar thing. It has some wet tests that distinguish between Al, Mg, Zn. I won't try to
write them here because I will get the details wrong. But a search might be useful.
[Edited on 4-3-2024 by j_sum1]
|
|
|
Bedlasky
International Hazard
Posts: 1251
Registered: 15-4-2019
Location: Period 5, group 6
Member Is Offline
Mood: Volatile
|
|
Alizarin turns Al(OH)3 red. Something similar can be done with Mg(OH)2 using titan yellow (red precipitate) or magneson (blue precipitate). Zinc can
be distinguished using ferrocyanide test (zinc ferrocyanide apparently quickly dissolves in NaOH and precipitate in dilute HCl). I also read about one
really simple method - Zn(OH)2 dissolves in excess NaOH or NH3, Al(OH)3 only in excess NaOH and Mg(OH)2 doesn't dissolve in either.
|
|
|
|








