Fery
International Hazard

Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
mononitrotoluenes (nitration of toluene)
Here important threads concerning this topic:
https://www.sciencemadness.org/whisper/viewthread.php?tid=29...
https://www.sciencemadness.org/whisper/viewthread.php?tid=28...
https://www.sciencemadness.org/whisper/viewthread.php?tid=10...
here some materials from Urbanski and Cumming:
  
according the literature I decided to synthesize at 2 mol scale and obtained 1 mol product (yield 50%).
calculations:
150 g 96% H2SO4
100 g 65% HNO3
Water content = 150x0,04 + 100x0,35 = 41 g
100 g of HNO3 is 65 g HNO3... 65 g / 63,01 g.mol-1 = 1,0316 mol
150 g of 96% H2SO4 is 144 g H2SO4
nitration mixture content:
HNO3 26%
H2SO4 57,6%
H2O 16,4%
2 mol of toluene = 184,3 g
---------- experiment -----------------
To a magnetic stirrer was placed a wide plastic dish with concentrated salt solution to which was added snow. To the freezing bath was inserted a 1 L
2-neck RBF into which was was placed 184,3 g toluene (2,00 mol, toluene molar mass 92,14 g/mol, density 867 g/l) and magnetic stirbar. To 1 neck was
inserted thermometer with its stem long enough to be submersed in toluene and to another neck was inserted dropping funnel with pressure equalizing
with a connector to which was attached a hose leading outside of building (you can also use 3-neck flask and takeoff adapter to the third neck to
which to connect the hose).
To 500 ml Erlenmayer flask was placed 200 g 65% nitric acid (130 g HNO3 = 2,06 mol HNO3 + 70 g H2O). It was placed into water-snow bath and to its
neck was inserted filtration funnel to reduce vapor escape. Through the funnel was dropwise added while rapid stirring and cooling 300 g 96% H2SO4
(288 g H2SO4 + 12 g H2O).
The nitration mixture has composition:
HNO3 = 130/500 = 26% wt
H2SO4 = 288/500 = 57,6% wt
H2O = 82/500 = 16,4% wt
Urbanski suggests the nitrating acid with composition 28 % HNO3 + 56% H2SO4 + 16% H2O and spent acid 1% HNO3 + 69% H2SO4 + 29% H2O. Temperature 60 C.
Reaction time 75 minutes. Ratio HNO3:toluene 1:1. Yield 97% theoretical. This is all from literature far from my laboratory results.
I started to add dropwise the 0 C cooled nitration mixture from the dropping funnel into rapidly stirred cooled toluene. The addition lasted 90
minutes and I managed to kept the T always below 20 C. According Cumming T 30 C is still OK. If you use crushed ice it is less powerful in cooling
than snow and you probably have to add reactants slower. Also at this my larger scale the cooling is less efficient as at smaller scale. There was
snow outside...
Then cooling bath was removed and reactor was rapidly stirred at room temperature for 30 minutes during which the T raised from 10 to 15 C.
Then it was rapidly stirred at warm water bath (aluminium pot, or glass vessel for the bath necessary), during 30 minutes the T raised to 50 C and at
this T it was let to react for further 2 hours while rapid magnetic stirring. At this stage there was a little red-brown NOx gas evolution which was
lead outside of building using a hose.
Using 500 ml separatory funnel I separated waste acid (bottom layer, higher density) and discarded it (it could be easily regenerated by boiling in a
beaker outside and obtaining back the concentrated H2SO4).
I washed the product with 200 ml of water (separation very slow, I had to let it in the separatory funnel overnight). I kept bottom layer (product,
density 1,16 g/l) and discarded upper aqueous layer.
Washed with 200 ml 2 % NaOH. Kept bottom phase. The upper water phase after washing was checked with universal indicator paper and it was alkaline.
Washed twice with 200 ml H2O. Kept bottom phase and discarded upper water phase. The last washing water phase was only pale yellow. The product phase
stayed milky (emulsion) even after sitting for half a day.
Product dried in 600 ml beaker using magnetic stirring and adding anhydrous CaCl2 in approx 2 g portions, and slightly heating to approx 50 C. RBF
placed at the top of beaker to prevent loses because of splashing on rapid stirring. First 2 portions of CaCl2 dissolved completely and the viscous
solution was pipetted out from the bottom of beaker and then fresh CaCl2 added. The third portion of CaCl2 did not dissolve completely and was let to
stir for 3 hours. The product now crystal clear, yellow-orange color, cca 220 ml (inaccurate measured only looking at the graduation of the beaker).
Decanted into 500 ml RBF from which vacuum distilled, at cca -80 kPa toluene distilled out, T in head 45 C (I did not expect that, so after toluene
distilled out I stopped vacuum distillation and replaced receiving flask with new one. The toluene fraction weighed 33,7 g. I guess a lot of it passed
further as 45 C is quite low (I saw some bubbling in the receiving flask too).
Then vacuum distillation resumed and vacuum increased to maximum of my 2-stage pump, collected everything in range 60-90 C, most of mononitrotoluenes
distilled in range 70-80 C and T of oil bath kept upto 140 C. At the end crystals of 4-nitrotoluene (m.p. 51,63 C) tried to clog the narrowest part
behind the condenser so I had to switch cold water cooling into warm water cooling (my heater system is set to 50 C but few degrees lost while warm
water travels through thermally insulated water tubes under the floor) and temporarily allow air to enter the apparatus so the clog was sucked back
into the apparats. Nitrotoluenes fraction weighed 144,6 g (1,01 mol, M=137,14 g/mol).
Residuum in distillation flask 32,5 g ??? dinitrotoluenes ??? - solidified when cooling to room temperature.
Urbanski claims ratio toluene : HNO3 1 : 1,01-1,05. I used ratio 1 : 1,03. Urbanski suggests T upto 60 C and Cumming upto 30 on addition of nitration
acid and upto 50 on heating - I kept upto 20 on addition and 50 on heating.
Urbanski claims yield 97%, my yield was 1,01 mol of mononitrotolenes from 2,00 mol of toluene. Also Cumming claims yield almost theoretical (148 grams
from 100 g toluene and 100 g HNO3 of density 1,44. I used commercial pure toluene of petrochemical origin, minimal purity granted 99%, freshly opened
new bottle of HNO3 claimed minimal concentration 65%, purity grade: pure, freshly opened new bottle of H2SO4 claimed minimal concentration 96%, purity
grade: pure.
Is here anyone else having only half of yield claimed in literature while strictly keeping reaction conditions?
The apparatus used for nitration, the dropping funnels is somewhat small, only 50 ml but I recharged it always with very cold 0 C next portion of
nitration acid mixture, the hose attached to the apparatus lead NOx outside of building but there was not yet NOx gas evolution observed yet.

The T on addition of nitration acid mixture into toluene was always kept below 20 C (later it was only 10 C near the end of addition).
Nitration is proceeding on addition...
 
After the addition the mixture was heated at water bath and the reaction reached 50 C, now NOx fumes very well visible. It was stirrer rapidly for the
whole time, here just to take a photo it was raised from the water bath to see the red-brown gas evolved.

Separation of mixture in 500 ml separatory funnel, here product in upper phase (density 1,16 g/ml) and waste acid as bottom layer.

Waste acid collected in beaker, it could be regenerated by heating outside, boiling out water and concentrating it back to 96% H2SO4 (I did not do
that)

Here second washing (first one was with water), this second washing with 200 ml 2% NaOH, it washes out phenolic compounds (sideproducts of oxidation
during the nitration process, to reduce side reactions the HNO3 should be in slight excess for the whole process - in my case it was, I used toluene :
HNO3 molar ratio 1 : 1,03, suggested ratio is 1:1,01-1,05). Aqueous layer in upper phase (later it was checked with pH paper and it was alkaline).
Product in bottom layer (density 1,16 g/ml).

The last fourth washing, now with H2O, upper water layer almost colorless, bottom product very milky (emulsion).

Drying with CaCl2 on magnetic stirred for a lot of hours, closing the top of beaker with round bottom flask to capture splashing on rapid stirring.

Vacuum distillation apparatus

Toluene distilled out at 45 C at pressure cca -80 kPa, toluene recovered 33,7 g.

Collected fraction 60-90 C, most of mononitrotoluenes distilled in range 70-80 C, vacuum level uknown, my vacuum gauge cannot measure these small
differences when compared close to few Pa, it is graduated in 2 kPa, my vacuum pump is 2-stage. Mononitrotoluenes weight 144,6 g = 1,01 mol, so from
2,00 mol toluene my yield was 50%.
         
[Edited on 12-1-2021 by Fery]
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
due to limitation these 3 pictures couldn't be posted in the previous post so I had to split them separately into this second post
near the end of distillation

clogged narrow part behind the condenser with 4-nitrotoluene (m.p. 51,63 C) I had to quickly switch cooling water from cold (10 C) to warm (50 C) and
stop vacuum by opening the vacuum control valve so the clog was sucked back into the apparatus and then I resumed the distillation after turning on
the vacuum pump and closing the vacuum control valve sucking air into the pump

end of distillation

[Edited on 12-1-2021 by Fery]
|
|
|
Mateo_swe
National Hazard
Posts: 560
Registered: 24-8-2019
Location: Within EU
Member Is Offline
|
|
Interesting, is mononitrotoluenes sensitive and can explode like 2,4,6-trinitrotoluene?
Not really regarding the experiment but when you distill solvents under vacuum what kind of vacuum gauge are you using?
A digital, mechanical round gauge or a U-shaped glass with mercury?
Doesnt the solvents damage the gauge and pump or du you use a trap to remove most things before it goes on to gauge and pump?
I have been looking at a digital gauge and a HVAC type mecanical vacuum pump but im worried solvents and vapours would ruin them if used without some
traps or a way to protect them.
Maybe i could try use a U-tube gauge with mercury if i have a digital gauge to calibrate the U-tube first.
I have an aspirator but no good way to display the vacuum and sometimes you want better than aspirator vacuum.
How do you do vacuum and measure it in your experiments?
|
|
|
Boffis
International Hazard
Posts: 1897
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Fery, another nice write up. I haven't tried this preparation for over 30 years but I don't remember getting such poor yield. Looking at your write
up though raises some issues. The published procedure asks for 100g of nitric acid SG 1.44 or 77% to 150g of conc sulphuric SG 1.84 but you have used
less concentrated nitric acid in the same ratio and claim the same nitration mixture composition. Or am I missing something?
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Meteo_swe - it is safe, I wanted to avoid dinitrotoluene formation but apparently cca 25 g of dinitro formed... I even tried to distill the dinitro
and it passed as a distillate, T in head 120 C and T of oil bath 160 C but I did not post that as it was not worth to do it and it could be dangerous
(but literature claims that explosions in manufacturers could be avoided when not heated to above 190 C (I was 30 C below).
My vacuum gauge is mechanical
https://www.sciencemadness.org/whisper/files.php?pid=651512&...
and the pump is 2-stage
https://www.sciencemadness.org/whisper/files.php?pid=651512&...
Well I decided to repeat the nitration, now during addition I kept T in range 0-5 C and then I allowed to raise T to 20 C but I wont heat it to 50 C,
I let it to rapid stir at the 20 C for 1 day or maybe 2 days, I started it 7 hours ago... I will see whether it could vacuum distill completely so no
dinitrotoluene formed at such low temperatures (but lit. claims no dinitro formed when T below 60 C) - I will see, practical experiment is always
better than literature.
Boffis you are right I used only 65% HNO3 but the nitration mixture was almost exactly the same as in Urbanski literature concerning explosives
production. The published procedure from Cumming also uses more toluene (100 grams of toluene for 100 grams of 77% HNO3) and I used ratio 1 mol
toluene (93 g) for 1,03 mol of HNO3 (100 g 65%) - in fact I was preparing at 2-fold scale (2 mols toluene + 2,06 mol HNO3). The 50% yield is strange
as toluene is easier to nitrate than benzene. I was more concerned about very likely dinitrotoluene formation although my nitration mixture was even
more diluted than in Cumming and I kept even lower temperatures during addition of nitration mixture into the toluene.
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
At least something from the mystery maybe solved - I tried to distill the "toluene" I recovered just to clean the condenser messed with nitroderivates
and have an image about the purity of the recovered compound (whether it could be reused) and first half of it boiled at 90 C and slightly more, but
not expected 110 C and also smelled like some aliphatic hydrocarbon (petrolether), not the well known toluene scent. I did not distill it completely
and stopped the distillation - I realized this is only waste of time.
I have to try toluene from another supplier and I'll see. But if this one is not claimed 99% but a mixture with something else, that could explain the
low yield and also the residuum after vacuum distillation (suspected to be dinitrotoluene) although dinitro should not be formed at 50 C and even with
already diluted nitration mixture... strange. If 32 g from 184 was recovered and this is not toluene then it is cca 17 %... but it still does not
explain the yield only 50 %.
[Edited on 15-1-2021 by Fery]
|
|
|
Boffis
International Hazard
Posts: 1897
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Ahh. That is interesting. I presume you recovered your toluene from some solvent type material. I wonder if the material contains petroleum spirits if
it could also contain something like amyl alcohol or butanol that are partly soluble in water or get oxidized to the corresponding acid.
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
better method of synthesis
https://www.oc-praktikum.de/nop/en/instructions/pdf/1001_en....
Attachment: 1001_en.pdf (179kB)
This file has been downloaded 456 times
I tried second attempt with the crappy "toluene", which was in this second experiment already almost consumed so it won't cause problems anymore (only
cca 2 ml left in the bottle). The above attached pdf uses 0,766 mol HNO3 for 0,5 mol of toluene so I can assume that I used similar ratio as my
toluene was cca 75%, at most 80%.
I used again the same amounts of "toluene" and acids. The same apparatus, but now much lower temperatures.
200 g 65% HNO3 (2,06 mol)
300 g 96% H2SO4
184,3 g toluene (2 mol if using pure toluene, in my case it was much less, cca 1,5 mol)
1 L RBF, 2 neck for reactor
1 L Erlenmayer flask for mixing nitration acid and keep it cold, the neck loosely "stoppered" with glass funnel.
Into 1 L Erlenmayer flask was poured 200g 65% HNO3, funnel was inserted into its neck (to reduce gas escaping at least a little, otherwise air
movement would release more of the choking NOx) and while cooling in water-snow bath 300 g of 96% H2SO4 was added dropwise while swirling in hand. It
was done outside.
I weighed empty 1 L RBF and put into it 184,3 g toluene while on the scale. Then added magnetic stirbar. Inserted thermometer into 1 neck and small 50
ml dropping funnel with take-off adapter into second neck (the dropping funnel should be small so the nitration acid is added into it cooled to 0 C so
it is dropping into the reaction as cold as possible, cold acid is added into the funnel cca 6-7 times during 2 hours). A hose was connected to the
take-off adapter to lead NOx outside of building (you can also use 3-neck RBF with the take-off adapter). Put water-snow-NaCl bath into plastic dish
to the magnetic stirrer and the RBF into the bath. The Erlenmayer flask with nitration acid was kept in a bucket with water-snow mixture.
Toluene was rapidly stirred and cooled down to 0 C, then I charged the dropping funnel and let the cold nitration acid to slowly drip into the
reactor.
The temperature was kept in range 0-5 C using snow-water-NaCl bath (you cannot achieve it using crushed ice, you need snow - big surface of
snowflakes). It was necessary to periodically drain a brine using a hose and add fresh snow and NaCl to the top and mix the cooling mixture. Also some
water/brine present is necessary as when mixing only snow with NaCl with lack of water/brine there is not enough liquid to transfer the cold to the
flask walls. The whole nitration acid was added dropwise during 2 hours while rapid stirring and then the snow was let to melt in the cooling bath so
the reactor was cooled further.
time +0h temperature 0 C, dropwise addition of cooled nitration acid started
time +2h temperature 5 C, addition finished, snow-salt-water bath let there to melt the snow
time +3h temperature 5 C, all snow melted
time +4h temperature 10 C
time +5h temperature 15 C (still let rapid stirring in the cold water bath, here the attached 1001_en.pdf says the nitration is done but I let it to
rapidly stir for totally 2 days, it is very likely an overkill but it does not hurt, maybe it was only waste of time and it did not improve my yield
significantly, I do not know)
time +6h temperature 20 C, bath removed (it already had room temperature so was useless, I did not expect too much unreacted reactant to start violent
reaction so late so I decided it is safe to remove the water bath)
time +48h temperature 20 C, rapid stirring stopped, total reaction time 2 days (maybe it was done in first 5 hours, who knows?)
Using 500 ml separatory funnel the crude nitrotoluene (upper layer, theoretical density 1,16) was separated from waste acid (bottom layer),
washed 2 times with 200 ml of water, upper water phase always clear and bottom nitrotoluene phase always milky (emulsion).
Washed with 200 ml 2% NaOH (after shaking and separation I checked pH of upper dark water phase using universal indicator paper, it was alkaline).
Washed 3 times with 200 ml water, the third washing was checked with universal pH indicator paper and it was neutral.
Dried by adding 5 g of CaCl2 and letting it sit for 2 days (now not rapid magnetic stirring) - it cleared up very nice.
Distilled from 500 ml RBF on oil bath, collected cca 30 ml of distillate boiling in range 90-108 C, cca 20 ml in range 108-120 C (temperature of oil
bath at the end 180 C) - this could be still toluene (but I don't believe as it lacked toluene scent, in my case it was perhaps something like
heptane) which vapors at the end were further heated up above its boiling point. I collected 45,0 g of colorless forerun. For you I do not suggest to
perform it at atmospheric pressure, I did it at atmospheric pressure in the second synthesis only because interested in separation of the shit present
in my "toluene 99% petrochemical" which in fact was something like 75-80% toluene + 20-25% of some undeclared shit.
If you repeat it I suggest to distill out unreacted toluene using a vacuum and oil bath at lower temperature. In my case in this second experiment my
dinitrotoluene residuum was much darker due to very high temperature unlike red-yellow in the first experiment when liquid distilled at cca 50 C from
oil bath cca 100 C.
Then using vacuum distillation I collected 147,9 g of nitrotoluene mixture with yellowish color, distillate boiling in range 65-95 C at maximal vacuum
of my vacuum pump (so in range of 30 C, most of the product distilled in range 68-85 C), at the upper boiling point range the distillate rate
significantly dropped cca 5-10 times. Do not distill further, in my previous experiment I was able to distill even dinitrotoluenes, but they will be
crystallized from the residuum in the flask.
Remainder in flask 24,7 g of dinitrotoluenes with brown color, in the previous experiment it was red-yellow.
I merged nitrotoluenes from both experiements together to process them further (144,6 g + 147,9 g).
As the weather forecast was -10 C during the night I put the nitrotoluene mixture outside in a stoppered flask. In the morining it was only -5 C but
there was a lot of crystalline mass so I filtered out the crystals using vacuum filtration on a sintered glass (the glass was let outside during the
night too to have the same low temperature). Then I quickly washed crystals on sinter two times with 5 ml of cold petrolether b.p. 60-70 C (which was
also cooled down to -5 C in a bottle let outside overnight).
Btw previously I tried to freeze mixture of mononitrotoluenes in my freezer at -18 C but it solidified completely (the 1001_en.pdf attached = link
posted here in the beginning suggests -20 C, but in my case it proven as a wrong way). Temperature -10 C would be even better but there is no such
forecast anymore soon.
If you perform filtration of crystals at room temperature I can imagine lower yield of crystals, try to use precooled funnel, schedule this synthesis
to winter time when outside temperature forecasted to -10 C and when there is a snow for cooling (crushed ice is not so much powerful due to much
lower surface when compared to snowflakes).
Now these steps are waiting for me to be done:
- recrystallization of dinitrotoluenes (remainders in distillation flask) from ethanol, it should give 2,4 dinitrotoluene.
- recrystallization of 4-nitrotoluene from methanol
- vacuum distillation of the liquid nitrotoluenes mixture on a column to obtain 2-nitrotoluene and perhaps then isolate some of para isomer from the
remainder in distillation flask too
So my suggestions:
Use the method in the attached 1001_en.pdf = the same as in the link.
Do not use the Urbanski suggested ratio toluene : HNO3 almost 1:1, but the molar ratio suggested here which is toluene : HNO3 = 1 : 1,5
Dinitrotoluenes are formed even on addition at 0-5 C and further nitration at 20 C in amount cca 10-15 %, when performing the addition below 20 C and
the further nitration at 50 C the dinitrotoluenes formed at cca 15-20%. Maybe using Urbanski suggested ratio almost 1:1 the dinitro are not formed?
Use pure toluene, my "toluene" was messed with some shit although claimed something else. I will never buy anything from the chem shop which sold me
this "toluene" - I could instead buy "toluene cleaner" from drugstore where I could expect mixture of toluene with something else and could distill it
at column.
I do not expect to perform nitration of toluene soon again as I have a lot of nitrotoluenes for further experiments.
I will post results of further purification and separation of isomers.
from left: 4-nitrotoluene crystals drying on filter paper, stoppered flask with yellow nitrotoluenes mixture partially depleted on para isomer,
remainder from distillation flask (dinitrotoluenes) in beakers, the darker on the right is from the second experiment when it was heated in upto 180 C
oil bath which I do not suggest (but lit. claims even 190 C is still safe)

|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
2,4 dinitrotoluene
Dinitrotoluenes are present in the remainder in distillation flask after vacuum distillation from which the most abundant isomer could be separated.
The remainder (33 g from first experiment and 25 g from the second) was dissolved in 100 ml of hot 95% commercial denatured ethanol (no need to boil,
it dissolves completely below boiling point).
On cooling down to room temperature, at first a dark oil separated, sunk to the bottom where it solidified and adhered to the bottom of beaker. Then
nice needle shaped crystals evolved on top of the dark solidified mass. These crystals were scraped while the dark bottom was not touched. Crystals
were filtered on sintered glass, quickly washed on sinter with little of ice cold ethanol (cca 5 ml), sucked well for few minutes and then were let to
dry on air for few days.
I melted them in small beaker and inserted thermometer showed melting temp. around 65 C which is good sign of 2,4 dinitrotoluene (m.p. 70C)
https://en.wikipedia.org/wiki/2,4-Dinitrotoluene
This isomer is the main of all dinitrotoluenes present there. Each of possible significantly present contaminants has m.p. at most 61 C
The second most abundant isomer present is 2,6 dinitrotoluene with lit. m.p. 56-61 C
https://www.chemicalbook.com/ChemicalProductProperty_EN_CB4223533.htm#:~:text=2%2C6%2DDinitrotoluene%20Properties,61%20%C2%B0C(lit.)
Possible contamination with mononitrotoluenes - melting points for the 2 most abundant are:
2-nitrotoluene m.p. -10,4 C https://en.wikipedia.org/wiki/2-Nitrotoluene
4-nitrotoluene m.p. 51,63 C https://en.wikipedia.org/wiki/4-Nitrotoluene
Cca 7,5 g of 2,4 dinitrotoluene obtained from each of my 2 experiments.
I will merge them together and recrystallize these 15 grams from ethanol again.
The mother liquor on cooling down to 0 C gave another crop of crystals and a little of them again when freezing to -18 C but I did not process them
further. Probably they could be processed the same way to give extra crop of 2,4 dinitrotoluene. Probably cooling down not to only room temperature
but even to 0 C could directly yield still pure enough 2,4 dinitrotoluene with somewhat higher yield? I was focused more on purity, not so much on
yield.
Mother liquor as well the remainder in distillation flask was much darker in the second experiment where I heated the nitrotoluenes in 190 C oil bath
to remove the shit contamination of my "toluene" - I was focused here to obtain more precise weight of the contaminant. In the first experiment where
I vacuum distilled it out at much lower temperature so some of it escaped further after collecting flask, the remainder in distillation flask and also
mother liquor after crystallization had better color. Though both crops of 2,4 dinitrotoluene had similar colors and similar approximate melting
points. Anyway I suggest to distill out unreacted toluene (in my case only contaminants without toluene) under
vacuum and do not heat dinitrotoluenes in such a hot oil bath as I did, not only because of color but also safety (lit. claims T upto 190 C
to be still safe).
[Edited on 26-1-2021 by Fery]
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
OK I finally have pure 2,4-dinitrotoluene from the remainders in distillation flask after vacuum distillation of mononitrotoluenes.
I recrystallized it from commercial denatured 95% ethanol+1% MEK+traces of denatonium benzoate.
for crude 15 g of dinitrotoluene use 100 ml of ethanol
cool down only to 20 C, you get cca 10 g of the product
recryst few times until m.p. 70 C (lit. value), my melting points are 69-70 C
the mother liquor obtained after crystallization at 20 C were further cooled down to 0 C, but these crops of crystals contained other isomers and
their melting points were lower than pure product, cooling down this mother liquor to -18 C in freezer yielded still a little of crystals and almost
nothing stayed dissolved in ethanol (I reused ethanol after distilling it and there was only little of solid remainder in the distillation flask after
distilling it out) - these isomers could be maybe oxidized with KMnO4 to dinitrobenzoic acid isomers and the acid decarboxylated to dinitrobenzene
use vacuum distillation to remove unreacted toluene, if you use high temperature instead of vacuum the product is darker, but its m.p. was still the
same 69-70 C, I believe that boiling with activated charcoal would improve its color
If anyone needs 2,4 dinitrotoluene, let me know (only trusted members and only small amouts which cannot be abused as an exposive).
the beaker with colored product is when I did not use vacuum for removing unreacted toluene but I heated the product to temperature IIRC like 180 C
instead
better colored product obtained when I removed toluene using vacuum pump
all samples have melting points 69-70 C (literature declares 2,4-dinitrotoluene m.p. 70C) the discolored product could be perhaps decolored using
activated charcoal
 
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
4-nitrotoluene
I repeat that the mixture of mononitrotoluenes was previously vacuum distilled (2-stage oil rotary pump, achieved vacuum level unknown but the
distillation range was 60-90 C, almost all mononitro passed in range 70-80 C). Dinitrotoluenes obtained as a residuum in distillation flask, mononitro
passed through condenser with finally clogging of vacuum receiving adapter (its narrow tube) at the end of distillation (use warm water for cooling at
the end).
Then the mixture of mononitroisomers was cooled to -15 C at which 4-nitrotoluene crystallized out. Cooling to -18 C in my case caused crystallization
of the whole mixture. Crystals sucked on sinter and washed with cold isohexanes mixture (b.p. 60 C) and let to dry. The crystals were stored in a jar
where they showed tendency to adhere to walls and to each other. I thought that this sticky behaviour is a sign of impure product.
Now the crystallization:
Approximately 50 g of p-nitrotoluene was dissolved in 100 ml of boiling methanol in a 250 ml RBF with reflux condenser attached (care, methanol is
toxic). Then cooled down to room temperature and further placed in a fridge with 4 C overnight. Crystals were sucked on sinter and washed twice with 5
ml of cold 4 C methanol from fridge. Air dried until the weight stable and let to stay on air for further 1 week, the weight did not change, but the
crystals still sticky, after 1-2 days they always lost their powdery attributes. These crystals are quite small and dense as they sunk to the bottom
or RBF and a lot of mother liquor was visible above them, I approximate the volume of bottom part with crystals in flask and volume of mother liqour
above them 1:1. Definitely not as some fluffy crystals which occupy the whole volume together with mother liquor.
weight 37,0 g
m.p. 51,0-51,5 C (lit. 51,63 C) which is very nice
The mother liquor was cooled in freezer overnight to -18 C, crystals quickly sucked on cooled sinter and washed 2 times with 5 ml of cold 4 C methanol
from fridge, air dried. These crystals are short needles and they behave sticky after few days too.
weight 6,5 g and melting point? Surprise! The same 51,0-51,5 C !
main crop 37,0 g (from fridge 4 C crystallization) on the right, second crop 6,5 g (from freezer -18 C crystallization) on the left

detail of needles from freezer (6,5 g crop)

detail of small dense crystals from fridge (37,0 g crop)

my 4-nitrotoluene is not snow white, both crops are very pale greenish
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
2-nitrotoluene, vacuum distillation using column
After months I finally had time, energy and mood to separate 2-nitrotoluene.
I repeat - from approx 290 g of mixture (purified by washing by water and dil. NaOH and vacuum distillation) I separated approx 50 g of 4-nitrotoluene
by freezing at -15 C (at -18 C it solidified completely) from which after recryst from methanol I obtained 43,5 g of 4-nitrotoluene (1st crop when
cryst. at 4 C and second smaller still very pure when further crystallizing mother liquor at -18 C). Moreover IIRC I recovered approx 3x 8 g of 2,4
dinitrotoluene from remainder in flask after vacuum distillation (various colors but nice m.p.).
After crystallization of part (approx 50 grams) of 4-nitrotoluene at -15 C, the remainder (mother liquor) mixture of 2-nitrotoluene + 4-nitrotoluene
(partially depleted) + little of 3-nitrotoluene (side reaction contaminant) weight 221,1 g was poured into 500 ml RBF, magnetic stir bar inserted,
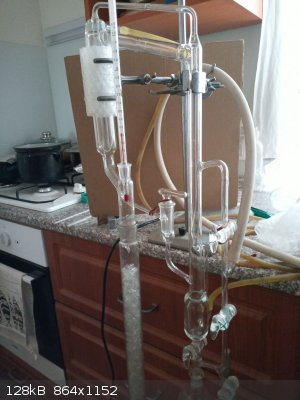
Hempel column with internal diameter 30 mm and lenght 600 mm packed with Raschig rings was attached and variable ratio reflux head with thermometer
assembled. Column insulated with approx 10 turns of newspaper and aluminium foil. Vacuum applied (2-stage rotary oil vacuum pump), cooling water
started, stirring started, heating started. 2 fans to cool vacuum pump also started to operate (such a long distillation essentially requires pump
cooling).
When distillation started, the initial temperature in head was 74 C under my vacuum pump (I'm unable to measure the vacuum level exactly, I do not owe
such presious manometer). After 15 minutes equilibrium at column established, temperature in head dropped to 71,0 C and did not decrease anymore so
distillate started to collect at a rate 1 drop into receiver : 5 drops returning back into the column.
Oil bath temperature 150 C (bath covered with aluminium foil to reduce heat loses).
Main fraction collected with boiling point range 71,0-71,5 C.
yield 157,5 g
Second fraction collected upto temp 75,0 C at the end close to 75,0 C very likely 3-nitrotoluene crystallized out at upper layer (bottom was still
very likely rich in 2-nitrotoluene)
29,2 g weight of the second fraction boiling 72,0-75,0 C (where solid m- or maybe even p- isomer crystallized on the top)
Some 4-nitrotoluene stayed on the surface of Rashig rings and column. I added 75 ml of methanol into apparatus and refluxed for 1 hour (with closed
stopcock so all distillate returned back into the column to wash out nitrotoluene into the distillation flask), then 25 ml of methanol removed by
opening the stopcock to flush and clean the glass.
here videos
https://youtu.be/8W3Yh4YU7fU
https://youtu.be/wInhNHZ6BqI
https://youtu.be/n4AzVz_68fI
assembling apparatus
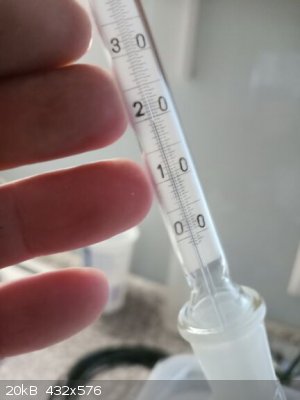
T in head rose to 74 C and later dropped to 71,0 C as equilibrium on column established

When T did not decrease anymore (71,0 C) the valve opened and part of the reflux taken off into receiver at a rate 1:5 (1 drop taken off, 5 returning
back into the column)

When most of the 2-nitrotoluene distilled out at 71,0 C the T started to increase, I stopped receiving main fraction at T 71,5 C and started to
collect second fraction (the receiver built in the head) while keeping the pure 2-nitrotoluene (main fraction) in the RBF (previously approx 60 ml
already removed from the receiving RBF in the middle of the distillation)

T still raising, now 72,5 C, still collecting the second distillate (I just wanted to as pure remainder in the distillation flask as possible with a
hope to distill out 3-nitrotoluene which is solid at room temperature so as 4-nitrotoluene)

At this point 3-nitrotoluene started to crystallize in the receiver part of the distillation head - on top. The cooling water made it to crystallize
while it managed to flow through warmer parts of the head. The bottom layer is richer in 2-nitrotoluene so liquid but the upper layer which seemed not
to mix with bottom layer solidified.
 
After finishing the distillation also crystals in the condenser part of the head clearly visible
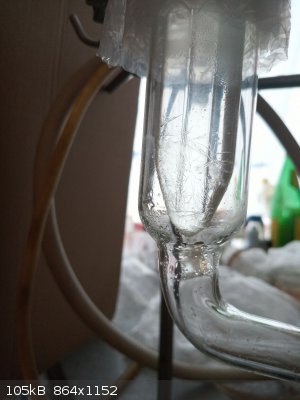 
|
|
|
Boffis
International Hazard
Posts: 1897
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Whooh; that's an impressive vacuum distillation rig! Have you tried running a TLC to test the purity of your products. I know its difficult with
volatile stuff like these but it does let you see what is present.
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Boffis, I didn't. I asked my friend and sc forum member Bedlasky to perform GC, he answered he would be able to determine all mononitrotoluene
isomers as well other possible contaminants like nitrophenols (I expect trace amounts), unreacted toluene, benzene (toluene contaminant, my "toluene"
was something else than declared 99% petrochemical toluene). I will send samples to him soon as I'm very curious how effective was that distillation
apparatus / setup / conditions.
Surprisingly boiling points at normal pressure differ much more than under vacuum.
2-nitrotoluene b.p. 222 C
3-nitrotoluene b.p. 232 C
4-nitrotoluene b.p. 238,3 C
In my vacuum distillation the 2-nitrotoluene distilled at 71,0 C and on 74-75 C there was clearly already solid isomer which could be 3- or
4-nitrotoluene (see pictures). Such reduction of temperature difference makes separation by distillation even harder and requires good column and good
head...
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
from the remainder in distillation flask after vacuum distilled out 2-nitrotoluene I obtained 12,0 g of 4-nitrotoluene m.p. 50,5-51,5 C by
crystallization from methanol (which I added to wash distillation apparatus) cooling down to 4 C (fridge overnight) and 3,9 g m.p. again the same
50,5-51,5 C by further cooling the mother liquor to -18 C (freezer overnight)
|
|
|
Fery
International Hazard
Posts: 1052
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
My friend Bedlasky performed gas chromatography analysis and the result is not good. But first good facts:
toluene 0
dinitrotoluenes 0
other organics 0,07%
H2O < 0,02% (must entered the sample from air while pouring from receiving RBF into storage bottle + the storage bottle is half full = bottle 250
ml from which 125 ml is the product).
now the contents of mononitrotoluenes:
83,7% o-nitrotoluene
3,35% m-nitrotoluene
12,88% p-nitrotoluene
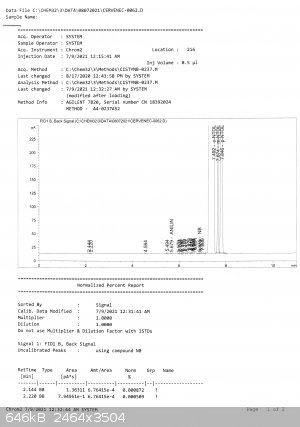 
it is very strange as the product distilled in range 71,0 - 71,5 C during few hours
I collected this fraction separately in receiving RBF below the stopcock.
Later the product distilling at incremental T upto 75,0 C during much shorter time as the main fraction (this second fraction weighed only 29 g while
the first one 157,5 g so the second fraction (with solid isomer of mononitrotoluene which could be meta or para or their mixture, orto is always
liquid at room temp) distilled in approx 5 times shorter time interval). The second fraction was stored in the receiver chamber part of the
distillation head (above closed stopcock) where it was visible that the distilling product solidified completely in top part of the receiver chamber
and in the tube (the cold cooling water made it to solidify in the top part of the chamber from which the crystallization process rose upward and
backflow into the connecting tube. Solidification made further distillation impossible. The bottom layers in the chamber stayed liquid (the
distillation T closer to 72 C than to 75 C presuming the layers did not mix too much and stayed in layers as entered the chamber).
So I do not know, whether the product could be some form of an azeotrope.
Or whether to use narrower Hempel distillation column, I used 3 cm diameter 60 cm length. Maybe at most 1 cm diameter (the condensation occurred at a
rate approx. 3 drops per second)? Or longer than 60 cm?
The packing was with glass Raschig rings, I have also copper mesh but the Raschig seems to be good choice.
The peaks in GC do not overlap. Bedlasky also performed GC with analytical grade o-nitrotoluene used for calibrating the GC:
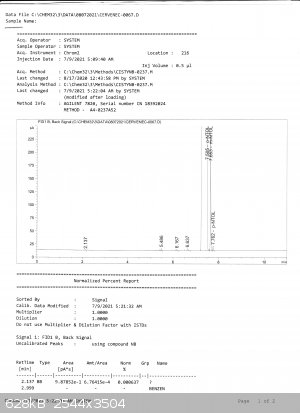 
here the result is
99,77% orto
0,20% meta
0,008% para
The nitrotoluene is usually only intermediate, so maybe I can take in consideration that it is only 83,7% and purify the product made from it
(o-nitrobenzoic acid, o-toluidine). But the b.p. difference among o, m, p mononitrotoluenes (222, 232, 238 C) is favorable for their separation much
more than for toluidines (200, 203, 200 C). Anyway under the reduced pressure the differences contracted almost twice as in the first vacuum
distillation (without column, separation of dinitrotoluenes) the distillation range for whole mononitrotoluenes was 10 C (IIRC 70-80 C), at atm.
pressure the theoretical range is 238 C - 222 C = 16 C).
And even if I perform second vacuum column distillation in the future I would separate again a fraction at boiling range of 0,5 C (see my thermometer
on pictures). So I'm afraid the second distillation wouldn't produce anything different. Air seemed not to enter the apparatus through some micro gap,
the temperatures were the same as when vacuum distilling without column (which separated dinitrotoluenes) and also the vacuum pump was the same
(meanwhile I just changed the oil). I used Korasilon vacuum grease for joints.
Pity that Magpie can't tell us his experience. In fact I made this experiment in honor of Magpie, he was the first one from us in the forum who did
it.
https://www.sciencemadness.org/whisper/viewthread.php?tid=29...
[Edited on 10-7-2021 by Fery]
|
|
|
|









