turd
National Hazard

Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
Tryptamine refusing to crystallize
For your amusement:
The decarboxylation of tryptophan to tryptamine has been discussed numerous times. The reference in this posting: https://www.sciencemadness.org/whisper/viewthread.php?tid=10... gives a nice overview on reaction conditions, catalysts, impurities. Very good
yields are expected for cyclohexenones (like naturally ocurring carvone). I am in the possession of oil of spearmint and oil of caraway, both of which
are supposed to be composed of > 50% carvone. Nevertheless I chose MEK as a catalyst since I didn't want to introduce a catalyst and impurities
with boiling points similar to the expected product. Yield is of minor importance since the tryptophan was obtained very cheaply. A very similar
procedure has been described by KrZ: http://www.erowid.org/archive/rhodium/chemistry/tryptamine2d... (only discovered it afterwards).
In a 200 ml round bottom flask with a condenser attached to a bubble counter filled with silicon oil, 10 g tryptophan was suspended in ~150 ml
tetralin and heated to a slight relux with vigorous stirring. 4 ml MEK were added until vigorous foaming set in(1). After ca. 45 min foaming slowed
down, and another 2 ml MEK were added. After a total reaction time of 1h30 the solution was not entirely clear(2), yet heat was removed and the
reaction cooled to room temperature. The insolubles were removed by filtration resulting in a sticky residue which was discarded. The tetralin was
stripped under reduced pressure (@65°C) and the dark red oil kept in a freezer for one week(3). The residue was distilled in vacuum (@165-166°C,
small forerun of tetralin) to give 3.1 g(4) of a very viscous pale yellow oil. The punchline: it refuses to crystallize! After two days in the
freezer the oil became turbid from small crystallization nuclei, but still it refuses to fully crystallize. Never seen anything like this, and I am
too lazy to crystallize from a solvent so will keep it as a curiosity as-is.
Notes:
1: The necessary amount of MEK will depend on the used flask and condenser, since a few ml MEK will always be located in the "head room" of the flask
and the lower part of the condesor.
2: In an experiment using caraway oil a clear solution was obtained after ~1h. But the colour was distinctly darker than in the MEK experiment,
hinting towards side products, possibly from polymerized terpenes or what not.
3: The residue stayed oily for one week in the freezer. After taking it out it crystallized in the next hour. Duh.
4: Yield seems mediocre, but is probably caused by an oversized distillation apparatus. I will have to organize a Kugelrohr one day.
|
|
|
gardenvariety
Harmless
Posts: 41
Registered: 19-1-2010
Member Is Offline
Mood: No Mood
|
|
The quick & dirty purification method is to extract into vinegar, basify, and freeze out the tryptamine, leaving short yellow crystals. I also
had relatively good luck with ethyl acetate/ethanol/water as a recrystallization solvent.
I didn't try MEK, but rather carvone and pulegone, both of which are cyclic enones, and both worked quite well.
[Edited on 12-7-2010 by gardenvariety]
|
|
|
peach
Bon Vivant
Posts: 1428
Registered: 14-11-2008
Member Is Offline
Mood: No Mood
|
|
Interesting turd, very interesting...
If you enjoy doing small scale experiments, might I recommend one of the following?

It's a short path distillation head. I think this would better suit your needs. The Kugelrohr is aimed more towards things that'll sublime and then
solidify in the next chamber.
That image is from United Glass Technologies, where they're $99 for a B14. It's essentially a tiny column, still head and condenser all in one and goes straight on
top of your tiny flasks for working with tens of ml's.
A lot of places sell variations on them, often with a few vigreux spikes in the column section or with the entire head encapsulated in a vacuum
jacket, for working with delicate or high BP materials.
Obviously, which such a short path, they're brilliant for doing analytical work on small samples, which'd end up stuck all over the walls of most
other glassware; even standard B14 columns, heads and condensers are too big for some of the work I like doing.
They go on the glassware in this kind of orientation;
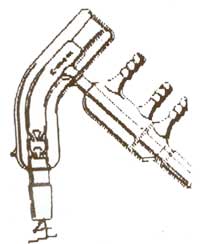
Sigma (Ace glass) do a version as well. I'd love one, but don't have the £350 - £450 (loads of $) or whatever they are around that;

They'd be great fun for doing lots of tiny tests. I could whip out a Gilson pipette for the super accuracy!
Stop complaining about the lack of crystals if you're not going to recrystallize, LAZY BOY! 
[Edited on 12-7-2010 by peach]
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Tryptamines can take a long, long, time to crystallize.....when they aren't in the mood.
Vacuum distilling them is no cinch either. High boilers.
I once synthesized an ounce or so of 2-Methyl- N,N-Diethyltryptamine on a lark.
For about two months, I was patient, but it never crystallized!
Eventually, to purify it, I vacuum distilled it and obtained a beautiful bright orange oil.
Once again, I waited, and waited, and waited.....But, no! It never crystallized!
Finally, exasperated, I added concentrated HCl to the oil. Bingo! Crystals of the hydrochloride salt formed instantly.
From there, isolating the pure amine was easy. I just neutralized the hydrochloride salt and crystals of the free amine formed.
Your Tryptamine might or might not, crystallize of its own accord. If you aren't going to prod it along, it could be a long wait.
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
try to dissolve it in a small amount of hexane and put it it freezer again, if your tryptamine freebase solidify it should come out as light-brown
crystals. Have you checked the purity of the crude product? Or as Zed points out, you might want to convert it to HCl salt. I would avoid using
concentrated HCl in this case, IMO it would be better to dissolve your oil in a small amnt. of ether and treat the solution with ether saturated with
dry HCl gas, then you could recrystallise it s HCl salt and liberate it after that if you want... Good luck turd! 
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Yup! Concentrated acids might be a bad idea for indoles that are unsubstituted at the two position.
|
|
|
xwinorb
Hazard to Others
Posts: 100
Registered: 9-8-2005
Member Is Offline
Mood: No Mood
|
|
I would suggest to do an A/B extraction instead of distilling out the tetralin.
Suggested procedure :
After filtering, add water, then add concentrated HCl dropwise, stirring, till ph is acidic. Try to keep the pH greater than 3. Better make it 6 - 7.
Separate the water layer, dry it on a hotplate on low heat.
You should have tryptamine hydrochloride.
I could not find the boiling points of both tetralin and tryptamine at the same pressure, but they sem too close. If you want to distill it, better to
separate the tetralin first with the A/B.
Tryptamine seem to have a reasonably low boiling point, it does not seem too difficult to distill with good vacuum. As already suggested, better to
use a shorth path.
Instead of the above, you could just extract the tetralin with slightly acidic water, again dry on the hot plate at low heat.
Nice post, thank you.
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
Thanks for the interesting responses. There seems to be more interest in tryptamine chemistry than I had anticipated! Of course the best way would be
(re)crystallization from a non-polar solvent, but since currently I neither have NaBH(OAc)3, nor NaBH3CN, nor LiAlH4, nor Hünig's base and
destructive distillation of quarternary ammonium salts doesn't look too attractive, I have time to see what it does. This morning I took it out of the
freezer and after 2 h most of it did crystallize, but there are still some uncrystallized parts. Now it's back in the freezer.
@gardenvariety, xwinorb: Yes, A/B is probably the way to go. But I tend to be too clumsy with such sensitive substrates, so I prefer to reduce the
number of manipulations to a minimum. I was thinking about washing the tetralin solution with a Na2CO3 solution to remove unreacted tryptophan, but
then opted not to.
@peach: These distillation bridges are cute.  I own a slighlty larger one for
semi-micro scale work. Unfortunately my smallest flask is quite large with 30ml. I own a slighlty larger one for
semi-micro scale work. Unfortunately my smallest flask is quite large with 30ml.  The problem with this distillation is that the oil is extremely viscous and I fear it would clog the condenser. Furthermore a huge advantage of a
Kugelrohr is that it generates a thin film, similar to rotary evaporators. This helps tremendously with high boiling oils like this one. Indeed take
off increased significantly when the stir bar started jumping around and spraying oil on the walls of the flask. Of course I will never be able to
afford a real Kugelrohr apparatus, but maybe I can improvise one from these Kugelrohr distillation bulbs. I've seen one, which would not make full
rounds but goes back and forth to avoid a complicated vacuum adapter.
The problem with this distillation is that the oil is extremely viscous and I fear it would clog the condenser. Furthermore a huge advantage of a
Kugelrohr is that it generates a thin film, similar to rotary evaporators. This helps tremendously with high boiling oils like this one. Indeed take
off increased significantly when the stir bar started jumping around and spraying oil on the walls of the flask. Of course I will never be able to
afford a real Kugelrohr apparatus, but maybe I can improvise one from these Kugelrohr distillation bulbs. I've seen one, which would not make full
rounds but goes back and forth to avoid a complicated vacuum adapter.
@zed: just out of curiosity: did you use reductive amination?
@sandmeyer: so far it crystallized to pale yellow crystals. But tryptamine seems to have multiple polymorphs and also crystal size has a big influence
on perceived colour. I will take a mp of the crystallized parts soon.
@xwinorb: The boiling points of tetralin and tryptamine are quite far appart. On my better pump, the tetralin came somewhere at 40-50° (didn't check)
and the tryptamine at 165°!
|
|
|
peach
Bon Vivant
Posts: 1428
Registered: 14-11-2008
Member Is Offline
Mood: No Mood
|
|
I've never seen a Buchi in person, but I'm skeptical about that joint being THAT hard to replicate. An overhead stirrer bearing does essentially the
same thing. There's also nothing special about their glassware. But it all costs a fortune. I just saw a non Buchi cold trap dewar for £10.50.
I found a lot of files about 4-hydroxytryptamines, but I'd like to have a sample of 5-hydroxy (serotonin) in my collection of amber bottles. One of
each of the neurotransmitters would be great. Think I'd start with serotonin, dopamine, GABA, acetylcholine and epinephrine (adrenaline).
I may be able to put my demethylated eugenol to use forming dopamine and adrenaline.
[Edited on 14-7-2010 by peach]
|
|
|
xwinorb
Hazard to Others
Posts: 100
Registered: 9-8-2005
Member Is Offline
Mood: No Mood
|
|
Partial pressure of tetralin
I found a PDF document that lists the partial pressure of tetralin at 1 mm Hg as 38 C. So, turd, looks like you are right.
Here is the link :
http://ntp.niehs.nih.gov/ntp/htdocs/Chem_Background/ExSumPdf...
I found also some Antoine coeficients for tetralin but I don't have the site info right now.
Looks like you have followed strictly the work up as described, and looks like it is OK to do so. I guess the A/B will do also.
In the Rhodium site they say the yield using MEK should be something like 85 %, if I remember it well. Is this a lot overstated, or what is not right
?
Maybe better to use something else rather than MEK ? ( as already posted here )
|
|
|
xwinorb
Hazard to Others
Posts: 100
Registered: 9-8-2005
Member Is Offline
Mood: No Mood
|
|
More exactly, the partial pressure of tetralin at 38 C is 1 mm Hg.
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
Yeah, in the end I got quite nice pale yellow birefringent crystals, but there still seems to be some tetralin in it (judged by smell), giving the
mass a disgusting mp (most at 100-105°). Recrystallization definitely is called for.
Quote: Originally posted by xwinorb  |
In the Rhodium site they say the yield using MEK should be something like 85 %, if I remember it well. Is this a lot overstated, or what is not right
?
Maybe better to use something else rather than MEK ? ( as already posted here ) |
Oh, I don't see why that kind of yield should not be achievable, especially if you do a longer reaction and chromatography. I probably lost most
during distillation. As I said the Trp was so cheap that I don't really bother about yields.
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
Wow, tryptamine(s) really do seem to have a live on their own. I repeated the above experiment with 150 ml tetralin, 20 g tryptophan and 2 g carvone
(fractionated out of spearmint oil) and had the opposite problem. On distillation the tryptamine immediately crystallized and clogged the
distillation bridge making it necessary to heat it with a bunsen burner (poor pump!). Stopped distillation after the temperature increased by 30°C
(due to fluctuating vacuum), but I'm sure I could have distilled some more. Got 8 g of slightly yellow tryptamine crystals.
Interesting: there was no carvone prerun, so I presume it reacted with tryptamine to the imine or THBCs.
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
Arise old thread!
Hey.
Need some newb help. I'm very unfamiliar with 2 things; salting amines and recrystallizations.
I have done as Student has in the decarboxylation of tryptophan in turpentine with spearmint oil as a catalyst.
What happened first is that I attempted the product, tryptamine, with no available means to test for it's purity or even formation. I left in the
freezer for a week. I reacted it with a alkyl bromide to get a hot mess.
Attempt 2 I have purchased some fumaric acid (this stuff does not like to dissolve in water!) in an attempt to salt it out. Problem is, I had no idea
what I was doing, and simply put some fumaric acid in water (I tried to first measure quantitatively the moles, but the dissolution caused errores
there) and pouring the acidified water into the solution of turpentine/tryptamine.
Against what I had assumed would happen - i.e. I would get a solid, I'm fairly certain the salt is just in the aq phase. SO my questions are:
1. Is this the correct procedure to salt the tryptamine, or could I have, for example, just added the fumaric acid to the original solution, stirred,
extracted [solids]? Or what would be the proper procedure in this case (I have searched but can only find people getting to the HCl salt)
2. Recrystallizations. I am only really familiar with solvent pair recrystallizations. What would be the ideal recrystallization for a salt in an
aqueous phase (assuming the stuff hasn't already degraded).
Or am I screwed
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by GreenD | Against what I had assumed would happen - i.e. I would get a solid, I'm fairly certain the salt is just in the aq phase. SO my questions are:
1. Is this the correct procedure to salt the tryptamine, or could I have, for example, just added the fumaric acid to the original solution, stirred,
extracted [solids]? Or what would be the proper procedure in this case (I have searched but can only find people getting to the HCl salt)
2. Recrystallizations. I am only really familiar with solvent pair recrystallizations. What would be the ideal recrystallization for a salt in an
aqueous phase (assuming the stuff hasn't already degraded).
Or am I screwed |
1. Obviously not. You generally don't precipitate salts from biphasic mixtures. Also, the first step in any such endeavor is to check the literature
if the salts is even known as a crystalline solid (Beilstein aka Reaxys is the best source for such info). Salts are best formed by finding a solvent
that dissolves the acid and the amine, but is a poor solvent for the salt. Fumaric acid is only soluble enough in lower alcohols, so you can try
ethanol or isopropanol. There is even a post that describes this.
2. The choice of solvents is based on estimations of the desired compounds properties and contrasting those with the expected properties of the
impurities (polarity, H-bond donors/acceptors, solubility, their amounts). Vogel's has a chapter on recrystallizations - read it.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
More success;
10g tryptophan added to ~125 ml Acetophenone.
Refluxed for 45 minutes. Solution turned from cloudy white to yellow to orange. After initial 45 minutes, heat lowered to slow simmer for an
additional 45 minutes. After 90 minutes total, solution was cooled so no more evaporation of acetophenone was observed and quickly capped air-tight
and left over night.
Solution gained slight red color. Small crystals forming at miniscus (!).
Distillation set up, and moderate heat applied. Lots of dissolved O2 came out. After 30 minutes at low pressure, boiling, but no condensation (worried
about pump!). Added more ice to capture bath, and salt. After 45 minutes, liquid started coming over. Bumping was very vigorous, some bumping fell
into flask. But, after catching ~10 drops condensate, spontaneous clear crystals form!
No melting pt apparatus or otherwise to find more properties. Must ingest.
Apparently tryptamine has lower bp than acetophenone. Large stalagmite in collection flask!
[Edited on 11-2-2012 by GreenD]
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
First of all, nice to read some real chemistry beyond speculation and crystallization.
But to be honest - that sounds a little bit fishy. I can't believe that tryptamine has a lower bp than acetophenone!? Was there lots of foaming during
reaction? Have you tried distilling pure acetophenone just to check your pump? What colour do the crystals have? What do they smell like? Acetophenone
has a mp of ~20°C - sure you haven't just frozen out acetophenone with your ice bath?
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
Turd, they definitely smell of acetophenone, and after they had sat out of the ice bath for approximately 1 hour they did appear to have some more
liquid. My house is a balmy 18°C.
I had read somewhere that the bp of tryptamine was 140° (I strongly believed that it was at 1atm) - which, yes, seems fishy to me as well since most
other tryptamines go up to around 400°!
Oh lord do I feel like an idiot! Don't know what I was thinking, getting crystal clear solids of tryptamine
[Edited on 13-2-2012 by GreenD]
[Edited on 13-2-2012 by GreenD]
[Edited on 13-2-2012 by GreenD]
|
|
|
turd
National Hazard
Posts: 800
Registered: 5-3-2006
Member Is Offline
Mood: No Mood
|
|
Hell, no! If you read the first post in this thread you will find that I distilled tryptamine at 165°C and not with the worst pump (probably ~1mbar
or less!).
I suggest practicing vacuum distillation with pure acetophenone: If you have trouble distilling acetophenone you probably have no chance distilling
tryptamine and should try one of the other purification methods. They are quite tricky too, but I had some success. It's essential to
meticulously follow the procedures!
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
I've got no problem distilling acetophenone, apparently!
I've searched this quite a bit and have found it difficult to get a straight answer; tryptamine HCl formation.
Addition of conc. hcl will most likely degrade the tryptamine, "acidic water degrades tryptamine". However, Tryp HCl salt is very stable.
I am wondering if adding conc. HCl to an acetophenone solution would precipitate Trypt•HCl? Since one, earlier, piece of advice suggested dissolving
tryptamine in ethanol (It is a great solvent for cleaning this nasty stuff) and adding conc. HCl.
Wouldn't this degrade the trypt? How is it done industrially (what I've been trying to find without success)
|
|
|
bahamuth
Hazard to Others
Posts: 384
Registered: 3-11-2009
Location: Norway
Member Is Offline
Mood: Under stimulated
|
|
You might try to preciptitate the tryptamine as the carbonate salt, by gassing with CO2 (guess one would need a little bit water to form the carbonic
acid). This is mentioned somewhere on the forum and was supposedly very clean.
Tryptamine degrade quicly in aquatic solutions strong in pH as you mentioned, so I would gas the reaction mix with dry HCl, or a part of it first to
test...
Any sufficiently advanced technology is indistinguishable from magic.
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
I've never seen how to prepare dry HCl, plus that scares me as I don't have a fume hood.
I read the carbonate salt once, very clever idea. I may try to (as was also recommended) salt out with a saturated fumarate/ethanol solution
|
|
|
Bot0nist
International Hazard
Posts: 1559
Registered: 15-2-2011
Location: Right behind you.
Member Is Offline
Mood: Streching my cotyledons.
|
|
I wouldn't recommend working with HCl without a hood. It preparation can be found in many chemistry books. I think you may find it in the SciMad
library, maybe Vogel. I imagine sulfuric acid + table salt + drying tube would do it. Remember that HCl is corrosive as hell and will attack almost
anything it touches. Sticking with gasses like CO<sub>2</sub> may be safer if you haven't built your hood yet.
Good luck, be safe.
U.T.F.S.E. and learn the joys of autodidacticism!
Don't judge each day only by the harvest you reap, but also by the seeds you sow.
|
|
|








