monoceros4
Harmless

Posts: 13
Registered: 30-8-2009
Location: Seattle, WA
Member Is Offline
Mood: No Mood
|
|
prepn. of stable non-K manganate or permanganate?
I was in the middle of a lengthy post about this when my browser blew it away so I'll content myself to a link to an even longer Google Doc describing my efforts, followed by an appeal. Has anyone here prepared a stable, well-crystallized permanganate salt
that's not potassium permanganate? Or a manganate? (BaMnO4 does not count.) Or even just a solution that keeps indefinitely? I can't seem to get
it right.
Basically, in pursuance of a microchemical test for potassium, I've been exploring the formation, in a test droplet under microscopic examination, of
characteristic crystals of slightly soluble potassium perchlorate. One description of the test can be found here in an old old journal of microscopy. One way to improve the test's sensitivity is to add a little permanganate to the test droplet,
whereupon the KClO4 crystals take it up in "solid solution" and are tinged pink or purple. Obviously the permanganate can't be KMnO4.
Basically I've failed to prepare any crystallized permanganate salt of any kind. I was aiming for ammonium permanganate because, despite its
unpleasant instability, it is also considerably less soluble than almost all other permanganates and therefore easier to crystallize. Nor have I
succeeded in preparing permanganate in solution that lasted at room temperature for more than a few hours. Manganates will do because they
can be used to prepare a permanganate solution in situ; indeed the old journal article I cite above recommends just that. But, again, all of
my manganate solutions have been unstable, even with refrigeration and considerable excess alkali, and the only stable salt I've prepared is a mixed
sodium sulfate-manganate decahydrate (and even that seems to keep well only in my makeshift 80% humidity box--in the open air it turns brown.)
Certainly I can do better?
"You must lead your opponent into a deep, dark forest where two and two make five, and the path leading out is only wide enough for one." Mikhail Tal
|
|
|
not_important
International Hazard
Posts: 3873
Registered: 21-7-2006
Member Is Offline
Mood: No Mood
|
|
Just a few quick comments.
One several occasions when you were acidifying you used HCl or sulfamic acid. Bad idea, permanganate will oxidise halide ions in acidic solution,
sulfamic acid may well be a target for oxidation as well. If you really need an acid solution stick to H2SO4 or H3PO4, perhaps HNO3 on a small scale.
Manganates are difficult to keep, the CO2 in air will cause the formation of MnO2 and permanganate, any dust is going to make some MnO2 as well.
Solutions of manganates need to be really alkaline to be anywhere near stable, too. Solutions of permanganates slowly decompose no matter what you
do.
On the small scale you are working with do your best to keep dust away. Use distilled water. Invest in a fritted glass filter unless you are positive
there are no organics in that fiberglass you are using.
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
A sodium perchlorate or perchloric acid solution would be much better for your purposes, because they are stable and the test will not interfere with
reducing substances.
|
|
|
monoceros4
Harmless
Posts: 13
Registered: 30-8-2009
Location: Seattle, WA
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by garage chemist  | | A sodium perchlorate or perchloric acid solution would be much better for your purposes, because they are stable and the test will not interfere with
reducing substances. |
If you'll trouble to read the Google Doc I linked to, you'll see that the test is in fact made with perchloric acid or a soluble perchlorate. There's
no question of its being "better" because it's already used. The permanganate is an addition to improve the limit of detection.
I knew that HCl was a bad idea but I was being hasty and using whatever I already had prepared. I had hoped that, in the absence of substantial free
acid, that oxidation of Cl- would be slow enough for the hastily-prepared solution to keep long enough to do something useful with it.
Permanganates do not oxidize sulfamic acid (Ind. Eng. Chem., 1938, 30 (6), 627-631) and I have at any rate confirmed this myself. A long while ago I
tried substituting recrystallized sulfamic acid for sulfuric acid in a titration with dilute KMnO4 and a "blank" containing only water and sulfamic
acid gave the permanganate endpoint color with half a drop of KMnO4 solution; nor did the color fade noticeably faster. As it turned out even the
unrecrystallized acid right out of the hardware-store tub did not reduce KMnO4.
In any attempt to make permanganates by acidifying manganates, sulfuric acid would be by far the best of the common acids, but I have none at the
moment and I was hoping to make do without having to go to the trouble of getting some.
Anodic oxidation of Na2MnO4 to NaMnO4 is well documented but so far I haven't worked up good equipment or technique for carrying out such an
oxidation.
Dust...is a problem here. It's a basement apartment, ventilation is poor, and I share my workspace with someone who uses an airbrush from time to
time. I haven't been doing anything special with my DI water other than occasionally boiling and cooling it when preparing certain solutions. I
didn't even bother with that here although perhaps I should have. The fiberglass cloth I'm using shouldn't be a problem because I cleaned it
exhaustively by long boiling in strong alkali, finishing off by extensive rinsing and then cooking it in alkaline 0.02M KMnO4 to confirm that it was
sufficiently clean.
"You must lead your opponent into a deep, dark forest where two and two make five, and the path leading out is only wide enough for one." Mikhail Tal
|
|
|
Paddywhacker
Hazard to Others
Posts: 478
Registered: 28-2-2009
Member Is Offline
Mood: No Mood
|
|
Barium manganate, http://www3.interscience.wiley.com/journal/114276064/abstrac... is said to be an easier reagent to use for oxidations than permanganate. A
verified preparation protocol would be useful to many people, I would think.
|
|
|
monoceros4
Harmless
Posts: 13
Registered: 30-8-2009
Location: Seattle, WA
Member Is Offline
Mood: No Mood
|
|
I did succeed in one experiment in making a small quantity of impure BaMnO4 by fusion of Ba(OH)2, MnO2, and KNO3. I could possibly improve on the
preparation if I tried it again. My aim was to prepare Ba(MnO4)2 by bubbling CO2 into a suspension of BaMnO4 in water. That failed, though.
Isn't barium manganate used as a pigment? Can it still be gotten that way?
I made the first trials of using the mixed sodium sulfate-manganate for the KClO4 microchemical test. The results so far have been disappointing.
It's especially frustrating because the first couple of slide tests looked promising but the photos I tried to take didn't come out. The formation of
permanganate was satisfactory but the KClO4 crystals did not take it up like the books say. The best I saw was scattered crystals with purple zones:
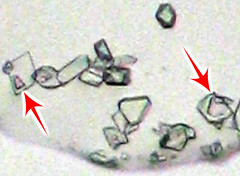
That was obtained by dissolving a few small crystals of the Na2(SO4,MnO4) in a large drop of aqueous 2% sodium perchlorate + 2% sulfamic acid, then
adding a droplet of this mixture to the residue left by evaporating a drop of dilute KNO3 (1 mg/mL in potassium). In no test with various
concentrations of KNO3 test-solution was any advantage seen with the permanganate-doped reagent. Moreover, most of the time it was hard to tell a
genuinely purple crystal apart from one that sort of looked purple because of the green and violet fringes you get with cheap achromatic optics. Oh,
well.
[Edited on 1-9-2009 by monoceros4]
"You must lead your opponent into a deep, dark forest where two and two make five, and the path leading out is only wide enough for one." Mikhail Tal
|
|
|
|











