| Pages:
1
2
3 |
Arrhenius
Hazard to Others

Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
Preparation of Iodomethane
Iodomethane is widely used as an 'active' methylating agent for the methylation of carbon, nitrogen, oxygen, sulfur and phosphorus. It can also be
used to prepare the Corey-Chaykovski reagent for cyclopropanation and epoxidation. Published procedures generally utilize phosphorus and iodine, which
are generally unavailable to the amateur. I have prepared iodomethane on 400mmol scale in 81% yield by direct halogenation of methanol.
Introduction
Alkyl halides can generally be prepared from the corresponding alcohol by treatment with the hydrogen halide.(ref. 1) The hydrogen halide may be
prepared in situ from its salt and a strong acid.(ref. 2) Phosphoric acid was employed to produce HI (Note 1):
H3PO4 + KI → HI + KH2PO3
While this equilibrium does not favor HI, it can be driven by consuming HI in the following dehydration reaction:
HI + ROH → RI + H2O
This reaction is entropically driven by the removal of the alkyl iodide.
Experimental
**Caution! Iodomethane is a probable carcinogen and is highly volatile!**
All reagents used were technical grade. I combined potassium iodide (66.4g, 400mmol), methanol (120ml, 3mol) and 85% phosphoric acid (175ml, 3mol) in
a round bottom flask and flushed the apparatus with argon (note 2). I performed a simple distillation at a relatively slow pace over a period of
approximately 3 hours (condensor at -5ºC). A clear product with a boiling point of 55-60ºC distilled over when the reaction mixture reached
90-100ºC. - the potassium iodide was entirely dissolved and the reaction mixture was red. I continued heating until the distillate temperature rose
to 70ºC, at which point the reaction should be stopped. I dilluted the distillate with 30ml of cold water and transferred it to a separatory funnel
(note 3). I removed the lower layer and dried it with anhydrous calcium chloride. A small amount of copper was added to stabilize the product for
storage (note 4).





Results
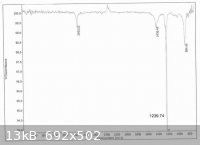
Crude iodomethane (45.83g) was obtained as a light red liquid in 81% of theory based on potassium iodide. Its IR spectrum is consistent with
published spectra.
Discussion
The product obtained in this scaled up procedure is of sufficient purity to use in subsequent reactions. The red color may be avoided by promptly
stopping the reaction when the distillate temperature rises above 65ºC. Fractional distillation of the product would also improve purity. Analagous
procedures may be used to prepare alkyl bromides in excellent yield; procedures are widely available online.(ref. 3)
Notes
1. Sulfuric acid should be avoided in the preparation of hydrogen iodide because it can oxidize the iodide ion.
2. Inert atmosphere is optional, but appears to improve purity and perhaps yield.
3. The obtained organic phase may be washed with sodium thiosulfate and/or sodium bisulfite to remove free iodine from the product.
4. Copper helps to absorb iodine resulting from the slow decomposition of the product. As with other alkyl halides, iodomethane should be stored in
the dark.
References
(1) Sidgwick, NV "The Chemical Elements and Their Compounds" p.1183 (available in sciencemadness library)
(2)Organic Syntheses, Coll. Vol. 4, p.323 (1963); Vol. 31, p.31 (1951). Link
(3) http://www.books-about-california.com/Pages/Experimental_Org...
[Edited on 6-7-2009 by Arrhenius]
|
|
|
CMOS
Harmless
Posts: 14
Registered: 27-2-2009
Member Is Offline
Mood: No Mood
|
|
nice work, i'm waiting for more.
why condenser is green??
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
PC coolant in recirculating system I suspect. Nice work Arrhenius. Perhaps a short fractionation column would help rid the orange/red colour?
[Edited on 6-7-2009 by DJF90]
|
|
|
Sedit
International Hazard
Posts: 1939
Registered: 23-11-2008
Member Is Offline
Mood: Manic Expressive
|
|
I was thinking that it was antifreeze.
Arrhenius is the H3PO4 lab grade or otherwise? I have been looking for an over the counter source but so far either highly dilute H3PO4 or navel
jelly(worthless) is all I have been able to find.
Nice work. I have been toying with a simular synthesis of Bromoethane but using H2SO4 and NaBr with mixed results due to oxidation of the HBr from
H2SO4.
Knowledge is useless to useless people...
"I see a lot of patterns in our behavior as a nation that parallel a lot of other historical processes. The fall of Rome, the fall of Germany — the
fall of the ruling country, the people who think they can do whatever they want without anybody else's consent. I've seen this story
before."~Maynard James Keenan
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
Very nice work. I can get a small quantity of 85% H3PO4 in the near future and will give isopropyl bromide a try by the same route.
Quote: Originally posted by Sedit  |
Nice work. I have been toying with a simular synthesis of Bromoethane but using H2SO4 and NaBr with mixed results due to oxidation of the HBr from
H2SO4.
|
I was having some spectacular failure at that same reaction using isopropanol. The quality of H2SO4 I have to use can't be helping either.
smuv must be magical: http://www.sciencemadness.org/talk/viewthread.php?tid=10758
[Edited on 7-6-09 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
All: The condensor is green because I use antifreeze and a circulating cooling bath. It gets to -10C on a good day, and the ethylene glycol won't
freeze. Looks high-tech too
DJF90: I suspect a fractional distillation would work quite well (as stated in the 'discussion'), but I'm not worried by the color. A highly pure
sample will start to turn redish-brown fairly quickly after the bottle has been opened a few times. I'm going to try some O-methylation shortly, I
suppose that'll be a good test.
Sedit: The phosphoric acid is technical grade. I know of one place to get it OTC, which is hydroponics stores. There are pH down products which are
conc. phosphoric with some phosphate salts in it. I highly doubt the salts will interfere with the reaction. As for ethyl bromide, I would suggest
you follow the prep. in the link at the end of my first post. I've used it to make upwards of 500g of EtBr in one run. Using H2SO4:H2O really
mitigates oxidation of bromine. I mean, the reaction is red, but the losses are negligible. I presume NaBr is OTC for you.
Unintentional Chaos: I've made quite a lot of ethyl bromide, and since sodium bromide is OTC (spa bromine source) I would suggest that you use
sulfuric acid and water to do the reaction. See the link above for the prep of ethyl bromide and follow that. Keep in mind that elimination to
propene may be a competing reaction if you heat too much or use too concentrated an acid catalyst. Purity of the sulfuric acid is not really an
issue. I've used brown drain cleaner, no problem.
[Edited on 6-7-2009 by Arrhenius]
|
|
|
Sedit
International Hazard
Posts: 1939
Registered: 23-11-2008
Member Is Offline
Mood: Manic Expressive
|
|
Thank you for the link Arrhenius. Yes NaBr is very over the counter in 52 gram packs for about $1.50 with 99% purity at Wal-Mart. It can be had
cheaper but I have yet to find larger containers with equal purity. I have been trying a slow addition of the H2SO4 in an ice bath to try to avoid
over oxidation but I feel H2O is a must if for nothing else to help dissolve the NaBr and bring it into solution.
As to not lead such a fine threed astray i'll pm you.
[Edited on 6-7-2009 by Sedit]
Knowledge is useless to useless people...
"I see a lot of patterns in our behavior as a nation that parallel a lot of other historical processes. The fall of Rome, the fall of Germany — the
fall of the ruling country, the people who think they can do whatever they want without anybody else's consent. I've seen this story
before."~Maynard James Keenan
|
|
|
Theophrastus_2
Harmless
Posts: 13
Registered: 6-10-2009
Member Is Offline
Mood: No Mood
|
|
Brilliant work arrhenius!
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Sedit: 58 wt% H3PO4 can be made from OTC materials. See
http://www.sciencemadness.org/talk/viewthread.php?tid=2923&a...
Arrhenius: In your top picture you show a tube leading to the RBF in the foreground. Is this for the argon? If so, did you use a continuous purge,
or just an initial flush?
[Edited on 7-10-2009 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
Magpie: Yes, it's an inert gas (argon) line. I purged it before I began heating, but that was it. On the two runs I made, it didn't seem to effect
yield, and perhaps only slightly effected the color of the product. With or without it, I think this is a robust method of preparing good purity
iodomethane. I can no longer do chemistry at home, but I sure would like to see someone take a stab at making allyl bromide *hint hint hint*. PM me
if you're interested.
If anyone repeats this synthesis, I would appreciate hearing about your yield, etc.
|
|
|
crazyboy
Hazard to Others
Posts: 436
Registered: 31-1-2008
Member Is Offline
Mood: Marginally insane
|
|
How easily can methyl iodide be substituted for other methylating agents such as dimethyl sulfate?
|
|
|
Sedit
International Hazard
Posts: 1939
Registered: 23-11-2008
Member Is Offline
Mood: Manic Expressive
|
|
You are turning methylating reagents into something generic. There are pros and cons of many things and it depends on the reaction at hand you plan on
using it for.
Knowledge is useless to useless people...
"I see a lot of patterns in our behavior as a nation that parallel a lot of other historical processes. The fall of Rome, the fall of Germany — the
fall of the ruling country, the people who think they can do whatever they want without anybody else's consent. I've seen this story
before."~Maynard James Keenan
|
|
|
crazyboy
Hazard to Others
Posts: 436
Registered: 31-1-2008
Member Is Offline
Mood: Marginally insane
|
|
| Quote: Originally posted by Sedit | | You are turning methylating reagents into something generic. There are pros and cons of many things and it depends on the reaction at hand you plan on
using it for. |
2,5-dimethoxybenzaldehyde from 2-hydroxy-5-methoxy benzaldehyde.
http://www.freepatentsonline.com/6670510.html
http://www.usdoj.gov/dea/programs/forensicsci/microgram/jour...
|
|
|
bahamuth
Hazard to Others
Posts: 384
Registered: 3-11-2009
Location: Norway
Member Is Offline
Mood: Under stimulated
|
|
My workup.
Methyliodide, Iodomethane.
To a 250ml RB flask in a cold water bath there was added 19g Methanol PA grade, 4.0 grams Phosphorus reagent grade.
Then there was prepared a condenser fit for reflux onto the RB flask and through it there were added 50.0 grams elemental iodine in portions so to not
cause boiling, the addition took about 5-7 minutes.
When the addition of the iodine were complete, the flask with the condenser still attached for reflux and cold water running through it, was left
stirring for about an hour or so before it was set on a hot water bath for gentle reflux for another half hour or so, before it was taken off the bath
to reach RT.
Quickly the flask was set up for simple distillation with the same condenser , with the distillate coming over into a 125ml sep. funnel cooled in a
Griffin beaker with icy cold water, again to ensure that no product escapes. The distillation was ended when no more came over to the sep. funnel.
The crude distillate, brown, milky, from iodine and water brought over by the MeI, was washed twice with 0,1M NaOH, top layer removed by decantation,
so it was clear and colorless, and once with RO water before it was transferred to a clean, dry RB flask with about 5-10 grams of anhydr. Calcium
chloride prills. This was left for 10 minutes or so to ensure all water was removed by the calcium chloride, and then set up for a final distillation
on hot water bath, with a brown preweighed flask for receiver. The RB flask was distilled to complete dryness for, yet again, ensuring no loss of
product. The product was perfect clear with no color.
This procedure should yield over 80%, my first try I got ≈47.5 grams which is ≈84.9%.
My second run, with three times the amounts listed above, yield rose to ≈89% (149.3 grams), as to why I do not know, perhaps a larger system
gives better yields.
Finally the product was stored over around .5 grams powdered Cu metal to act as stabilizer in a brown black painted bottle with Teflon lined cap.
From 200 grams of iodine, 197 grams of MeI came over, with ≈223.7 grams as 100%, giving an overall yield ≈88%.
BTW, red phosphorus can be obtained from vendors who sell "bulk" crystal iodide on ebay, ask if they got a website...
[Edited on 3-11-2009 by bahamuth]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Interesting work up. Why do you wash with NaOH? This seems strange to me; I would work up the reaction using a sodium thiosulfate wash followed by a
brine wash. This should remove all the residual iodine, and most of the water in your product. Drying can then be commenced over CaCl2, although I
would probably use potassium carbonate myself. But nice prep, and nice yeild.
|
|
|
bahamuth
Hazard to Others
Posts: 384
Registered: 3-11-2009
Location: Norway
Member Is Offline
Mood: Under stimulated
|
|
I washed with 0.1M NaOH because I found this old student assignment on the net somewhere, cannot remember where now though, but here is the original
writeup:
129. Preparation of Methyl Iodide from Methyl Alcohol and Phosphorus Iodide (Sections 49, 204).
- In a 200 cc. round-bottomed flask place 15 grams of methyl alcohol and 3.2 grams of red phosphorus.
Have ready a reflux condenser with cork attached.
Place the flask in cold water and add in small portions at a time 38 grams of iodine;
the addition should take about 10 minutes.
If the contents of the flask begin to boil, attach it to the reflux condenser;
when reaction ceases add more iodine.
Finally attach the flask to the condenser and let it stand for at least 4 hours (preferably over night).
Distil through a condenser from a water-bath, as long as any liquid passes over.
The receiver should be placed in cold water as methyl iodide is very volatile.
Wash the distillate by decantation with a dilute aqueous solution of sodium hydroxide
until the lower layer is colorless, and then once with water.
Separate the methyl iodide carefully from the water using a separatory funnel,
and transfer it to a small distilling flask.
Add about 10 grams of anhydrous calcium chloride.
Stopper the flask, place a cork over the end of the side-arm, and set aside until the liquid is quite clear.
Place a thermometer in the flask, connect the latter with a condenser, and distil from a water-bath.
Note the boiling-point and weight of the methyl iodide.
Calculate the theoretical yield from the iodine used (why iodine?),
and the percentage of this obtained.
Methyl iodide boils at 44°, and has the specific gravity 2.27 at 15°.
The yield in the experiment should be about 80 per cent of the theory.
Note. - An excess of the alcohol is usually taken in preparing alkyl halides by the method illustrated above.
The phosphorus and iodine are used in the proportions necessary to form phosphorus tri-iodide, PI3.
Also did this workup synthesizing propyliodide with a little less yield, I think, could not find my papers just now to confirm.
[Edited on 3-11-2009 by bahamuth]
|
|
|
entropy51
Gone, but not forgotten
Posts: 1612
Registered: 30-5-2009
Member Is Offline
Mood: Fissile
|
|
Nice job bahamuth, but for some of us that would be a waste of iodine and phosphorus, both of which are watched, especially on ebay. MeI can be made
using NaI and H3PO4 or gaseous HCl, which are more accessible and safe to order or make. www.hms-beagle.com was also selling MeI recently.
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
That procedure using NaOH is in Reference 3 from my initial post. In my opinion, the phosphoric acid method still represents the safest, most
economical way to prepare iodomethane.
|
|
|
Anders Hoveland
Banned
Posts: 208
Registered: 15-6-2010
Member Is Offline
Mood: No Mood
|
|
Iodomethane is known to react with anhydrous AgClO4 dissolved in benzene to make AgI precipitate and methyl-perchlorate, which is likely to
spontaneously explode if separated from a solvent. Using IodoEthane would, then give Ethyl Perchlorate, which is discussed elsewhere on this forum.
The precipitation reaction is rather slow, and AgClO4 hydrate is impossible to dry by heating (AgClO4 will explode).
|
|
|
crazyboy
Hazard to Others
Posts: 436
Registered: 31-1-2008
Member Is Offline
Mood: Marginally insane
|
|
How should one go about disposing of the reaction mixture and cleaning the distillation equipment in a safe way?
EDIT: never mind, sending it to HazMat disposal.
[Edited on 18-10-2010 by crazyboy]
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
Whoa really? I would just chuck it down the drain.... frankly. There's not much alkyl iodide in it, and the acid will be diluted to harmless
concentrations, and the methanol has a very short environmental half life.
|
|
|
crazyboy
Hazard to Others
Posts: 436
Registered: 31-1-2008
Member Is Offline
Mood: Marginally insane
|
|
| Quote: Originally posted by Arrhenius | | Whoa really? I would just chuck it down the drain.... frankly. There's not much alkyl iodide in it, and the acid will be diluted to harmless
concentrations, and the methanol has a very short environmental half life. |
Honestly I dilute just about everything and toss it down the drain. But methylating agents are something else, wouldn't want to mutate the sea life
and all.
|
|
|
peach
Bon Vivant
Posts: 1428
Registered: 14-11-2008
Member Is Offline
Mood: No Mood
|
|
| Quote: | Carcinogenicity in mammals:
Methyl iodide is listed under California Proposition 65 (1986) as a chemical known by the state to cause cancer or reproductive toxicity based on
evaluative studies performed in the 1970s.[18] It is considered a potential occupational carcinogen by the U.S. National Institute for Occupational
Safety and Health (NIOSH), the U.S. Occupational Safety and Health Administration and the U.S. Centers for Disease Control and Prevention.[19] The
International Agency for Research on Cancer concluded based on studies performed after methyl iodide was Proposition 65 listed that: “Methyl iodide
is not classifiable as to its carcinogenicity to humans (Group 3).” As of 2007 the Environmental Protection Agency classifies it as "not likely to
be carcinogenic to humans in the absence of altered thyroid hormone homeostatis," i.e. it is a human carcinogen but only at doses large enough to
disrupt thyroid function (via excess iodide).[20] However this finding is disputed by the Pesticide Action Network which states that the EPA’s
cancer rating "appears to be based solely on a single rat inhalation study in which 66% of the control group and 54-62% of the rats in the other
groups died before the end of the study". They go on to state: "The EPA appears to be dismissing early peer-reviewed studies in favor of two
nonpeer-reviewed studies conducted by the registrant that are flawed in design and execution."[21] Despite requests by the U.S. EPA to the Pesticide
Action Network to bring forth scientific evidence of their claims, they have not done so. |
I have phosphoric, phosphorus, KI and iodine, but noted the wiki article mentioning;
| Quote: | Methyl iodide can also be prepared by the reaction of methanol with potassium iodide, catalyzed by acid:
CH3OH + KI + H2SO4 → CH3I + K2SO4 + H2O
The reaction is carried out at low temperature and the water generated in the reaction is trapped by excess sulfuric acid so the reaction is not
reversible. The generated methyl iodide can be distilled from the reaction mixture. |
I have also seen this appearing elsewhere.
There a few oddities about the wiki article, such as;
- The acid in the formula is not a catalyst, it's a reactant.
- Also from the formula, the sulphuric is not acting as a drying agent. On the contrary, it and the methanol are the source of the water on the
right hand side.
Adding concentrated sulphuric to iodide salt and warming the two up was the route by which iodine it's self was first isolated. I posted photos of
me rerunning this original isolation in this thread, when I accidentally made iodoacetone towards the end of it.
I suspected the wiki note was edited based on hearsay.
Arrhenius has already demonstrated the preparation with phosphoric acid, and bahamuth has shown it with phosphorus. The other method is with DMS, not
a healthy thing to be using. Even if the yields were quantitative, I would still prefer the phosphoric option.
So I thought I'd try this supposed sulphuric mention.
I carried it out in much the same manner as Arrhenius, but swapping phosphoric for 98% sulphuric.
| Quote: Originally posted by DJF90 | | Interesting work up. Why do you wash with NaOH? This seems strange to me; I would work up the reaction using a sodium thiosulfate wash followed by a
brine wash. This should remove all the residual iodine, and most of the water in your product. Drying can then be commenced over CaCl2, although I
would probably use potassium carbonate myself. But nice prep, and nice yeild. |
Any moisture around the PI3 method will cause it to decompose, with one of the products being hydrogen iodide; a gas down to -34C. Any that escapes
may, in some percentage, end up in the resulting distillate.
I don't think a column is all that worthwhile if it causes much extra effort. To ensure all of the product is over, it's best to allow it to go up to
the BP of methanol, as Arrhenius suggests.
If there is any iodine in the hot flask, enough of that will make it's way over, column or not, that it'll benefit from a thiosulphate rinse.
Which I did, and it does a great deal to clean the result up.
I also rinsed with brine.
| Quote: | | In my opinion, the phosphoric acid method still represents the safest, most economical way to prepare iodomethane. |
Agreed. The only real competition for it is with phosphorus. Which is a lot more effort to get hold of and adds some potential health risks that are
not present with phosphoric. It also means measuring out iodine it's self; much messier than KI.
----------------------------------------------------------------------------
Experimental:
I took the copypasta equation from wiki and scaled it to 25ml.
The methanol and KI were combined and cooled in the freezer, with the sulphuric sat next to them. I prepared a salt / ice bath and sat the flask in
there, adding a squirt of the acid and giving them a swirl until it was all in, then set up a simple distillation.
Distillation began with a colourless, low viscosity liquid coming over at the BP for methyl iodide. The temperature was gradually rising in a
continuous band towards that of methanol.
After approximately 1 to 1.5ml of colourless liquid had been collected, a dark brown began appearing within the still and was soon crossing over. I
continued the distillation until the temperature fell back down from around 70C - all volatiles removed.
I had started with 5.8g (approx. 7ml) of methanol. The theoretical yield of methyl iodide would therefore be 24.8g (approx. 10.9ml). My actual volume
was 7.3ml of something blatantly contaminated.
Retaining the volatiles in the distillation tube and using a disposable pipette, I washed with 3ml x 2 water to remove the methanol. Then again with
3ml x 2 of thiosulphate, making the second more concentrated as it did not seem to be budging the staining too quickly as it was. The second wash
entirely removed the stain. I finished by washing with 3ml of brine.
The layers separate very nicely throughout, but I left this to stand for approximately an hour whilst I tidied up and preweighed a test tube
(14.24533g). Returning, I noted the volume of the suspect iodide as being 2.7ml. I then withdrew the base layer, placing it into the preweighed tube
and noting that less approx. 0.1ml remained in the distillation tube.
I remeasured the mass of the storage tube and found it to be 19.68300g. Giving a mass change of 5.43767g for a volume of 2.6ml, and so a density of
2.09g/ml, with that of methyl iodide being 2.28g/ml. The density obtained would be correct if the volume was 2.8ml. As I made no attempt to
standardize the temperature and the graduations on those tubes are likely not super accurate, that's reasonably close (8% off). My sample may also
have a trace of moisture or methanol left in it.
The yield from methanol (using sulphuric in place of phosphoric) is a terrible 22%.
I cleaned a small length of copper wire in sulphuric, then under a running tap, gave it a dry and dropped it in with the methyl iodide. Which shall
live in the fridge for now until I can find something more suitable to store it in.
Tidying up:
The sulphuric has oxidised most of the iodide to iodine. The glass is full of it. I knocked and rinsed it back out, removed the (rock solid) lump of
potassium sulphate from the flask, dumped it all into a big beaker, filled it up with water, stirred it all to get the sulphate dissolved, then ran it
all through a filter paper to recover the (so large it's actually useful) mass of iodine.
Yep
It does work. It works badly.
There is probably some way to increase the yield with sulphuric but I doubt it's worth it.
I have made ethyl iodide in the past and found mentions of preferably leaving it to reflux overnight (bahamuth has posted an example including that).
When I tried ethyl iodide by PI3, only refluxing for a few hours, I did note a low yield. With the method described by Arrhenius, similarly, it may
increase the yield a little if it's left to go overnight.
For anyone else preparing it, the point Arrhenius made to distill up to the BP of methanol is important. As you can see, I had just over 1ml of
colourless methyl iodide prior to the brown beginning to appear and the temperature rising, yet I have double that after washing by allowing the
distillation to continue upwards.
<a href="http://img835.imageshack.us/i/img2142sq.jpg/" target="_blank"><img src="http://img835.imageshack.us/img835/1962/img2142sq.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img268.imageshack.us/i/img2143rt.jpg/" target="_blank"><img src="http://img268.imageshack.us/img268/965/img2143rt.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img688.imageshack.us/i/img2144w.jpg/" target="_blank"><img src="http://img688.imageshack.us/img688/5613/img2144w.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img805.imageshack.us/i/img2148f.jpg/" target="_blank"><img src="http://img805.imageshack.us/img805/1927/img2148f.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img809.imageshack.us/i/img2149v.jpg/" target="_blank"><img src="http://img809.imageshack.us/img809/5721/img2149v.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img412.imageshack.us/i/img2150i.jpg/" target="_blank"><img src="http://img412.imageshack.us/img412/5465/img2150i.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img576.imageshack.us/i/img2155cj.jpg/" target="_blank"><img src="http://img576.imageshack.us/img576/5789/img2155cj.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img35.imageshack.us/i/img2159kj.jpg/" target="_blank"><img src="http://img35.imageshack.us/img35/6223/img2159kj.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img84.imageshack.us/i/img2160y.jpg/" target="_blank"><img src="http://img84.imageshack.us/img84/8929/img2160y.jpg" alt="Free
Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img52.imageshack.us/i/img2162j.jpg/" target="_blank"><img src="http://img52.imageshack.us/img52/9098/img2162j.jpg" alt="Free
Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img215.imageshack.us/i/img2163i.jpg/" target="_blank"><img src="http://img215.imageshack.us/img215/2316/img2163i.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img502.imageshack.us/i/img2165y.jpg/" target="_blank"><img src="http://img502.imageshack.us/img502/9957/img2165y.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img406.imageshack.us/i/img2166g.jpg/" target="_blank"><img src="http://img406.imageshack.us/img406/4153/img2166g.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img825.imageshack.us/i/img2167e.jpg/" target="_blank"><img src="http://img825.imageshack.us/img825/7955/img2167e.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img850.imageshack.us/i/img2168f.jpg/" target="_blank"><img src="http://img850.imageshack.us/img850/7420/img2168f.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img822.imageshack.us/i/img2169l.jpg/" target="_blank"><img src="http://img822.imageshack.us/img822/2046/img2169l.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img508.imageshack.us/i/img2176a.jpg/" target="_blank"><img src="http://img508.imageshack.us/img508/7066/img2176a.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img189.imageshack.us/i/img2177to.jpg/" target="_blank"><img src="http://img189.imageshack.us/img189/3664/img2177to.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a><br>
<a href="http://img268.imageshack.us/i/img2179q.jpg/" target="_blank"><img src="http://img268.imageshack.us/img268/2425/img2179q.jpg"
alt="Free Image Hosting at www.ImageShack.us" border="0"/></a>
That's the phosphoric, phosphorus and sulphuric variations tried. And some sweet team action. Hands up who's
doing DMS?
[Edited on 3-1-2012 by peach]
|
|
|
entropy51
Gone, but not forgotten
Posts: 1612
Registered: 30-5-2009
Member Is Offline
Mood: Fissile
|
|
Methyl iodide can also be made by gassing a saturated solution of KI in methanol with HCL gas.
I've not been able to find the ancient reference in which I found this method, but I have used it and it does work. I'd been thinking that I should
post something on it since it does not seem to be generally known.
|
|
|
Sedit
International Hazard
Posts: 1939
Registered: 23-11-2008
Member Is Offline
Mood: Manic Expressive
|
|
Peach if you do not have phosphoric acid perhaps you could use your Sulfuric and react it with Toluene to make TsOH. This is a non-oxidizing acid that
should work rather well in this synthesis. Its solid so its easy to weigh and handle.
Its funny this thread was just brought up because I just came in from setting up an electrochemical cell with EtOH, NaBr and NaOAc to see if I can do
a Kolbe coupling to produce Methyl bromide. Just another day of me tossing things together and playing around. Once of these days I swear I will take
chemistry seriously
Knowledge is useless to useless people...
"I see a lot of patterns in our behavior as a nation that parallel a lot of other historical processes. The fall of Rome, the fall of Germany — the
fall of the ruling country, the people who think they can do whatever they want without anybody else's consent. I've seen this story
before."~Maynard James Keenan
|
|
|
| Pages:
1
2
3 |