| Pages:
1
2 |
len1
National Hazard

Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Sulphur Monochloride, Dichloride, and Thionyl Chloride - Illustrated Practical Guide
This article is part of a series of Illustrated Key Syntheses in Chemistry which I intend to post, with the purposes and guidelines outlined in the
thread of the same name in the General Chemistry section. I shall attampt to ensure that all the relevant information is contained in a single post.
If sufficient reason arises for something to be changed or edited, I shall edit the entire post.
Aim
This article presents a three-step procedure for producing crude thionyl chloride SOCl2 at 62% yield based on SO3. The product is of constant 76-78C
b.p. range but nevertheless contains a significant impurity of sulphur chlorides as evidnced by IR. The SOCl2 is obtained in the third step by
oxidation of sulphur chlorides with SO3 (itself reduced to SO2), the preliminary steps thus being the synthesis of crude sulphur monochloride S2Cl2
(98% yield), and sulphur dichloride SCl2 (quantitative), which for lack of effect on yield and purity of the SOCl2 have not been individually
separated (in the sense of separating a product of pure stoichimetry from the S/Cl2 system - see theory).
Thionyl chloride is a mixed-acid anhydride whose main role is in effecting chlorine substitution of OH groups in organic derivatives such as alcohols
and acids, where the substitution reaction with HCl does not proceed due to instability of the intermediate carbion
R-OH + SOCl2 -> R-Cl + SO2 + HCl
R-COOH + SOCl2 -> R-CO-Cl + SO2 + HCl
This results in alkyl halides and acid chlorides, respectively. The equilibrium is favourably shifted by gas evolution, which has the additional
benefit over such reagents as PCl3, that only gaseous by-products are formed.
Acid chlorides of low boiling point - close to the 77C of SOCl2, such that can not be separated by fractionation, can nonetheless be separated from
the SOCl2 by destroying the residual of the latter using a variation of the second equation above
HCOOH + SOCl2 -> CO + SO2 + 2HCl
Thionyl chloride has a fairly strong dehydrating capacity due to its reaction with water (R=H in the first equation above)
SOCl2 + H2O -> 2HCl + SO2
This leads effectively to water elimintation rather than alkyl halide formation for tertiary alcohols - for the latter purpose however HCl can be
used.
SOCl2 can be used to form anhydrides directly from organic acid salts without isolating acid chlorides as intermediates
2R-COONa + SOCl2 -> R-CO-O-CO-R + SO2 + 2NaCl
This reaction is heavily exothermic and proceeds more readily than the the two stage process of an acid chloride and the anhydrous salt.
The sulphur chlorides also have a range of uses. S2Cl2 can be used to form acid chlorides
R-COOH + S2Cl2 -> R-CO-Cl + 1/2SO2 + 3/2S + HCl
and anhydrides
2R-COOH + S2Cl2 -> 2R-CO-0-CO-R + 1/2SO2 + 3/2S + 2HCl
as well as mono- and di-sulphides with aromatics
S2Cl2 + Ph-H -> Ph-S-S-Ph + 2HCl
S2Cl2 gives condensation products with phenols - a good demonstration of which has been provided (with great timing) by Klute http://www.sciencemadness.org/talk/viewthread.php?tid=10365.
Reaction with olefins yields sulphide addition products. Such reactions should not be carried out by the amateur chemist, however choosing propylene
(as a less controvertial example) one gets
2CH3-CH=CH2 + S2Cl2 -> CH3-CH(Cl)-CH2-S-CH(Cl)-CH3 + S
Reacting S2Cl2 with carbon disulphide yields carbon tetrachloride
CS2 + 2S2Cl2 -> CCl4 + 6S.
Reacting the dichloride SCl2 with an excess SO3 yields the (rather stable) anhydride of chlorosulphonic acid (a rather vicious compound by all
accounts)
SCl2 + 3SO3 -> S2O5Cl2 + 2SO2
The latter reaction has had a direct impact on the present experiment whose aim it to generate SOCl2 and hence minimise the yield of S2O5Cl2 (b.p.
148C) - the SCl2 had to be used in excess.
Findings
A 62% yield of crude SOCl2 based on SO3.
The crude SOCl2 has a constant b.p. of 76-78C, but IR (as well as a light yellow colour) reveals significant impurities which can not be
separated by distillation (see theory).
S2Cl2 obtained by feeding Cl2 at a rate of 400ml/min into liquid S at 220-250C gives a yield of 98% based on sulphur (using b.p. as end-point,
see theory).
Procedure leaves no hard-to-clean S deposits in reaction flask or condenser - only milligram quantities of soot left in the flask (cleaned with
ethanol).
Continued feeding of Cl2 into cooled S2Cl2 (else reaction heats up to 40C) at 400ml/min achieves SCl2 stoichiometry for the S/Cl2 system with
no free chlorine in the exhaust. Chlorination can continue up to 10% beyond this stoichiometry with no undisolved chlorine.
S2Cl2 like all sulphides is poisnous, with an all-pervasive hard-to-get-rid-of smell resembling sewer gas. It dissolves plastics and latex - a
stray drop on a glove generated pulpable heat at that spot within seconds.
The crude SOCl2 smells of SO2 and sulphur chlorides due to impurities of the latter (as in the commercial tech product).
Theory
The S / Cl2 system
Despite what is often stated sulphur was not found to react with Cl2 in the cold. There is a good reason for this - the start of reaction is an
unstable process. If S is present in a particularly reactive form, then a spot reaction generates sufficient local heat to fan the process as the
reaction is exothermic
2S + Cl2 -> S2Cl2 delH = -60 kJ/mol
There is no need to rely on this - the sulphur only needs to be heated externally to 80-100C to ensure complete absorption of Cl2 fed into a 250ml
flask at a rate of 400ml/min (1 drop 15%HCl into TCCA per 2 secs).
Despite the exothermicity of the reaction S2Cl2 is quite an unstable compound, which decomposes into its elements at 300C. In addition both S and Cl2
are soluble in S2Cl2 to a great extent (corresponding to stoichometries of about S5Cl2 at the one extreme, and S2Cl5 at the other) - it is clear
therefore that it is difficult to differentiate precisely the amount of mixture and chemical compound in the S/Cl system - and indeed for many
purposes (such as subsequent reaction with SO3) such a differentiation is not important. Clearly a substantial amount of the sulphur and chlorine
must be chemically bound owing to the completely different physical properties of the product from the reagents. Reaction of the bound elements with
SO3 will have the effect of 'denuding' the non-bound elements in solution (this was indeed observed) which will promptly react by Le Chattliers
principle to form more of the compound.
For the purpose of enabling a separation of S2Cl2 as part of the present synthesis, the reaction temperature was maintained at 220-240C which ensured
evaporation of the S2Cl2 as soon as it formed, while the Cl2 being fed into the sulphur meant negligible SCl2 formed. This arrangement also has the
additional benefit that impurities such as ash presented in the sulphur stayed in the reaction flask.
Oxidation of sulphur chlorides with SO3
In its simplest form this reaction is usually written as
SCl2 + SO3 -> SOCl2 + SO2
where the sulphur in SCl2/SO3 is oxidized/reduced to the +4 level. However SCl2 is even more unstable than S2Cl2 and the pure compound (b.p. 59C
obtained by slow distillation from PCl3) decomposes in a matter of days back along its formation reaction to attain the equilibrium
S2Cl2 + Cl2 -> 2SCl2
If we consider a variation of the above reaction, such as used by Greisham-Elektron, where SO3 is reacted at 75C (in a disporoportionation reaction)
with the monochloride while passing in a stream of chlorine
S2Cl2 + SO3 -> SOCl2 + SO2 + S
2S + Cl2 -> S2Cl2
then multiplying the bottom reaction by two, adding, and cancelling the S, gives
S2Cl2 + Cl2 + SO3 -> 2SOCl2 + 2SO2
so we see that the SCl2 reaction is nothing but a time-reordered version of the S2Cl2 reaction, where in the former case the Cl2 is already present,
either dissolved or reacted with the S2Cl2, while in the latter case the lacking Cl2 is added during the reaction. For this reason no step
was taken to isolate a pure SCl2 product in this experiment.
Reactions of thionyl chloride
One of the major uses of thionyl chloride is to effect the substitution of the hydroxide group for chlorine in organic chemistry, especially in
situations where HCl can not perform this feat
R-OH.....................->...R-Cl
R-CO-OH...............->...R-CO-Cl
...............+ SOCl2..->...............+ SO2 + HCl
H-OH.....................->...H-Cl
H-CO-OH...............->...H-Cl + CO
One of the great advatages of this over other chlorinating agents such as PCl3 or S2Cl2 is that only gaseous byproducts are formed, while the last
reaction can be used to destroy any left over thionyl chloride reagent without the need for a distilation.
The reaction proceeds via the formation of the intermediate ester with the anhydride SOCl2
R-OH + SOCl2 -> R-O-SOCl + HCl
which then proceeds with the elimination of SO2, the ester being unstable. However for certain classes of R, notably aromatics, the ester can be
isolated.
Thionyl chloride can also be used in acid anhydride synthesis, which is formally a dehydration, with normally a positive enthalpy
2R-CO-OH -> R-CO-O-CO-R + H2O
by adding the thionyl chloride reaction with water (negative enthalpy)
SOCl2 + H2O -> SO2 + 2HCl
the water cancels out between the two equations above and we get an overall process with negative enthalpy
2R-CO-OH + SOCl2 -> R-CO-O-CO-R SO2 + 2HCl
The above equations of course represent thermodynamic considerations (it matters not whether water actually forms at the molecular level) - a
necessary but not sufficient condition for the reaction to proceed - but it enables us to compare the strength of potential reagents for anhydride
formation. For example, S2Cl2 which is claimed in the Chemical Encyclopedia to perform a similar transformation, has the corresponding reaction
S2Cl2 + H2O -> HCl + 1/2SO2 + 3/2S
whose enthalpy is about 30kJ/mol more negative than the SOCl2 reaction. It thus provides greater thermodynamic impetus for anhydride formation -
there is however the inconvenience of colloidal sulphur being produced.
Purification of SOCl2
In addition to the desired SOCl2 product the following by-products can be present at the end of the third step
S2Cl2
SCl2
S(a)Cl(b)
SO2Cl2
Cl2
SO2
SO3
To complicate matters, the SOCl2 itself decomposes into these by-products during the distillation
2SOCl2 -> SO2 + Cl2 + SCl2
The gases SO2 and Cl2 are a problem being soluble in the liquid products (Cl2 greatly so, while with no substantial frothing being evident during the
third step, SO2 must also be very soluble), but like all gases become much less soluble at the boiling point of the liquid, indeed the smell of SO2
did not become evident until the contents were refluxed. The SO3 has a sufficiently low b.p. (44C) that most of it escapes uncondensed (heavy mist
evolved during earlier part of distillation). This can also be minimised by using excess of sulphur chlorides. SO2Cl2 has a b.p. very close to 77C
for SOCl2, while SCl2 has a b.p. of 59C. The latter can be substantially removed by adding elemental sulphur to effect conversion to the
monochloride, which with the b.p. of 138C can be easily separated from the SOCl2 by fractional distillation. However as detailed in the US patent
3155457, the SCl2 reforms from the S2Cl2 in the lower half of the distillation collumn
S2Cl2 -> 2SCl2 + Cl2
which escapes the collumn, and contaminates the product with an equilibrium mixture of the S2Cl2/SCl2 system. The method suggested for purification
(Vogel etc) is to use quinoline to neutralise the acids, while toluene or linseed oil reacts with the sulphur chlorides, in which they are
preferentially soluble.
Method
All operations described below were carried out in a fume hood, wearing a gas mask, latex gloves, and with a
drought established in the laboratory.
Ambient temperature in the lab 20.
Step 1 - formation of S2Cl2 from the elements
This procedure is borrowed from Schlessiners "Inorganic Laboratory Preparations" - with the procedure in Brauer judged unsuitable. 100 grams of
garden-grade sulphur were charged into a 250ml flask, into which dried Cl2 from the TCCA generator was fed at a rapid rate (about 5 bubbles/sec = 1
drop 15% HCl per 2 secs onto TCCA in 1L flask). The Cl2 was dried with a small dehydrated CaCl2 plug, and then bubbled into H2SO4. The H2SO4 bubbler
was deemed suitable as contamination with H2SO4 spray in this case is of little relevance, as the product will be sulphonated in any case. A water
cooled condenser led to a 250ml collecting flask - with the exhaust gases vented to a NaOH solution. When the sulphur melted the Cl2 delivery tube
was lowered almost to the bottom of the flask (as otherwise it would not introduce the Cl2 below the S surface once a substantial amount of product
had evaporated) and the walls of the flask were soon covered with at first yellow, and later red drops. See picture below
<IMG src="http://www.sciencemadness.org/scipics/Len1/SOCl2_fig1.JPG">
After an initial lag (when the S2Cl2 product dissolved in the sulphur) drops of yellow/orange fluid started to gather in the receiver (about 1 drop /
2 sec). A 40% duty cycle on the controller ensured a steady flask wall IR temperature of 220C. Testing with conc. NaCl solution showed no Cl2 was
passing the flask uncombined. After about 400ml of HCl had been added (90mins) the boiler flask was bone dry and completely empty, save for a few
black particles of soot. The receiver flask was half-full of an orange solution, which as soon as no distilate was coming over immediately started to
redden from the surface, and feeding proressively down, due to formation of red SCl2. See picture below.

The generator was stopped and flask weighed at this stage - total product weight was 208 grams, giving a yield of 98% based on sulphur. Considering
almost no Cl2 escaped, this provides very strong support for S2Cl2 being the formula for the component boiling below 220C.

Step 2 - dissolution of Cl2 & formation of SCl2.
S2Cl2 has very high density (about 1.7) so 71.9 grams of the product from step 1 were weighed into a 100ml flask placed in an empty vessel to allow
cooling of the contents (this increases the solubility of Cl2 as with all gases). A small amount of steel wool (< 100mg) was dropped inside the
flask. The outlet of the reaction flask was protected from moisture with a CaCl2 plug and an H2SO4 dried stream of Cl2 was passed in as before at the
rate of 4-5 bubbles/sec.

The colour of the liquid started to redden almost immediately - at the end it looked almost black (due to depth of colour, thin streaks were still
red). While there were some perturbations on the surface of the sulphur chloride initially, no gas was seen to exit the flask. The contents warmed
to about 36C, when water was added to the pan which cooled the flask to 20C. At this stage the reagent surface was calm despite furious bubbling in
the H2SO4 drier. Some perturbations began to appear on the sulphur chloride surface again when the weight gain of the flask corresponded to a
stoichiometry of SCl2 - this could be used to determine when to stop the addition. Some red condensed drops of SCl2 also became evident in the upper
parts of the flask, carried there by the Cl2 flux, and showing that as opposed to S2Cl2 the SCl2 compound has a low b.p. At this stage the weight of
the product had risen to 109.8 grams so 37.9 grams of Cl2 had been absorbed. The stoichiometry of the solution therefore corresponds to SCl2.002.
The iron wool was extracted from the solution, and was found to be completely uncorroded - except for a dark-black surface sheen.
Step 3 - oxidation of SCl2 to SOCl2
The procedure from Brauer was employed for this with a few modifications. Essentially the SCl2 is placed in a cooled vessel attached to a reflux
condenser and SO3 is added. Although some sources quote direct dripping of 65% oleum into the SCl2 reagent, this introduces substantial useless water
into the reaction, while evaporation of 65% oleum is wasteful of SO3 (the b.p. of 25% oleum is already 140C) hence 90% SO3 made using the SO3
Illustrated Guide, was evaporated into the SCl2. Due to the low b.p. of this material a heat gun was applied directly to the U-tube containing the
SO3. Brauer recommends using H2SO4 to prevent ingress of moisture into the refluxing mixture - however this is quite inappropriate as very little SO2
is evolved immediately and the temperature inside the vessel fluctuates enough (initial heating followed by natural cooling) to create the real
possibility of suckback of the H2SO4. A CaCl2 tube was employed in its stead. The cooling vessel consisted of an iron pan placed on a hot-plate -
this way after the addition of SO3, which produces heating, is complete, reflux could be maintained by external heat. The setup is seen below.

There is the danger here that if the SO3 at the bottom of the U-tube is heated to boiling before the SO3 at the top had melted, a major part of the
U-tube contents can be ejected under pressure into the flask containing the SCl2. Hence the tube is heated evenly, strating from the top, letting that
section melt, before working further down, until all contents are molten and at the bottom of U-tube. Soon the SO3 starts boiling, and dripping into
the SCl2 containing flask. Drops of the condensing SO3 as well as SCl2 start refluxing and the condenser insides become yellow due to Cl2 and SCl2
vapour being released by the heated mixture. Sulphur also starts precipitating on the flask walls, inside the condenser, and inside the flask, slowly
sinking in a lump to the bottom. This is what Brauer meant by 'the contents of the flask might solidify'. The excess chlorine liberated started
eating at the precipitated sulphur on the walls (in a repeat of the S2Cl2 formation reaction), so we in effect have a perfect demonstration of the two
equations from the theory section, showing that this is the main reaction mechanism
S2Cl2 + SO3 -> SOCl2 + SO2 + S
2S + Cl2 -> S2Cl2

Once sufficient SCl2 refluxed and SOCl2 formed there was some dripping of this back into the SO3 vessel, where together with excess SO3 they combined
into a deep blue liquid compound - see picture below. As the chlorosulphonic anhydride S2O5Cl2 is supposed to be colourless, I am not sure as to the
composition of this substance formed when SO3 is present in excess.

Once all the SO3 had been added, the mixture was refluxed at 80C for half an hour. Upon cooling some of the sulphur had cleared. The mixture was
then distilled. As soon as the temperature is raised the receiver and condenser are filled with free chlorine, which has the effect of dissolving any
sulphur deposited on the flask walls. A key point Brauer doesnt mention, is that even with the SCl2 being in excess not all SO3 is converted, so the
first distilate is SO3 boiling at 44C and not condensing very well. The distilate must be vented outside. Next the thermometer jumps to 59C and a
red liquid comes over - this corresponds to the characteristics of SCl2. The temperature then rises to 75C while a few ml of reddish liquid comes
over. Most of the fraction comes over as light yellow drops in the range 75-95C range, which redden as free chlorine passed over it. Sufficient S
was then added to convert the sulphur chlorides to S2Cl2,

and the product distilled through a lagged collumn. There was no lower b.p. compound this time, as soon as some liquid had started coming over the
temperature shot up to 76C and did not rise to more than 78C before no more distilate came (the mantle was operated at 25% duty). The sulphur, and
some terribly smelling highly sulphur chlorides stayed in the boiler. The distillation was probably still a little fast, about 2 drops/sec. A total
of 59 grams of a light yellow liquid smelling of SO2 and S2Cl2 had been obtained from 64 grams of SO3 added, the 109 grams of SCl2 being in excess.
The yield based on SO3 is then 62%.

The SO3 oxidation was conducted in two different ways. The first time, an H2SO4 drier at the condenser vent was employed, which, as the reaction is
prone to suck-back as mentioned above, at the end of the reflux allowed about 15 ml 95% H2SO4 to drip into the reactor flask - which had the effect of
converting the 64 grams of free SO3 already added into 65% oleum. This instantly dissolved all sulphur deposits, and formed a second layer on top of
the sulphur chlorides. Curiously this had the effect of actually increasing the yield of SOCl2 by about 10% compared to a second run where a CaCl2
drying tube was used and 100% SO3 added.
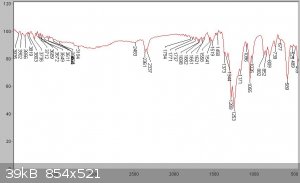
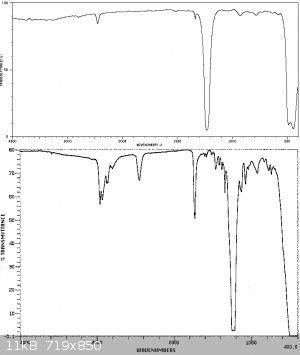
An IR spectrum of the product is shown below compared to the following IR spectra in the literature. As can be seen substantial impurities are still
present in the SOCl2 due to the difficulty in purifying this compound of sulphur chlorides. The reference spectra manifest this problem too - being
sufficiently different from each other. This is characteristic of a product exhibiting some instability, so its compoisition varies with time and
with different sources. It should also be mentioned that due to traces of SO3 present in the sample, which reacts with atmospheric moisture to form
H2SO4, a reaction with the KBr window of the FTIR sample card produced some spotting of the KBr window, which could have led to some of the artifacts
observed. The product should be washed in quinoline to remove the acids and distilled from linseed oil as mentioned in Vogel, however due to a lack
of the former this process has not been carried out at this stage.
It is interesting to note that the reflux condenser and flasks were all covered with colloidal sulphur at the end of the reaction. On disconnecting
the apparatus they fumed strongly due to residual SO3 combining with moisture from the atmosphere. Over the course of several hours the oleum
so-formed completely cleaned all vessels by oxidizing the sulphur according to the reactions
S + 2H2SO4 -> 3SO2 + 2H2O
Conclusion
Thionyl chloride at 62% yield based on SO3, a constant b.p. but with substantial impurities still present has been obtained. The impurities and
yellow colouration are a common problem with technical grade SOCl2 and require several fractionations from quinoline and linseed oil to eliminate.
S2Cl2 and SCl2 have also been obtained at 98% yield in the preliminary steps.
[Edited on 21-4-2008 by len1]
[Moderator Edit Feb 2019 – reformatting image links]
[Edited on 12-2-2019 by j_sum1]
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
See Brauer for a much cleaner SO3 oxidation of sulfur chlorides.
Or, see Kyrides for FAR superior method, the ZNCl2 catalyzed reaction of SO2 (not SO3) with phthaloyl chloride.
I have posted both of these previously.If you use the FSE and search on Kyrides you will find it straightaway.
SO2 is a lot more user friendly and easier to make than SO3.
The purification of crude SOCl2 contaminated with sulfur chlorides like the product above, is by boiling with linseed oil then with quinoline.
Phthaloyl chloride is likewise easily prepared from benzotrichloride and phthalic anhydride and catalytic ANCl2, as described in same article. You get
like 98% yield of both phthaloyl chloride and benzoyl chloride. The ZnCl2 catalyst is even reusable.
The patent literature teaches that the oxidation of sulfur chlorides can be done with 65% oleum rather than neat SO3, Brauer simply uses 65% oleum as
a convenient source for neat SO3 rather than as a reactant per se.
The same patent states that the oxidation is done under sutogenous pressure and Cl2 must be passed in to suppress the side reaction which forms
pyrosulfuryl chloride (thus wasting sulfur chlorides and oleum).
However IMO anytime there are safer and practicable alternatives which give high yields of cleaner products, I would pass on the use of SO3 which is
life threatening and not just hazardous.
The reaction of phthaloyl chloride with SO2 (Cat.ZNCl2) is just such a reaction. Phthalic anhydride is no problem to obtain. You yourself demonstarted
the preparation of halogenated toluene, of which benzotrichloride is the last.
The resulting SOCl2 needs little purification.
No dangerous SO3 or oleum is employed.
[Edited on 19-4-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
| Quote: | Originally posted by Sauron
See Brauer for a much clner SO3 oxidation of sulfur chlorides.
|
As mentioned in the text, the method is from Brauer, so Im not sure what 'cleaner' method youre referring to. I have made some improvements, also
mentioned there.
| Quote: | | The purification of crude SOCl2 contaminated with sulfur chlorides like the product above, is by boiling with linseed oil then with quinoline.
|
I also mention this phrase from Vogel in the text - except your ordering is the wrong way - first quinoline, then linseed.
The method of Kyrides you have been preaching very regularly - no need to shout louder, it hasnt gone amiss. Its an interesting reaction - easy to
describe, not so easy to achieve. Try making some benzotrichloride and you'll see why!
[Edited on 19-4-2008 by len1]
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
I find it very inspiring what you're able to achieve with very basic reagents plus good apparatus, research, and hard work. You've shown that few
materials are off-limits to the determined amateur, even if you have to start from the elements to get there.
PGP Key and corresponding e-mail address
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Actually I buy my benzotrichloride, so I needn't be overly concerned with chlorinating it up from toluene, I leave that to you.
For that matter I buy my phthaloyl chloride, as well, but I did purchase 4 liters of benzotrichloride just so I can replicate Kyrides anytime I like.
I suggest you reread Kyrides as he has important things to say about the limitations and failures of SOCl2 in preparation of anhydrides, whereas you
so glibly present it as if it were a general reaction, which it is not.
And the author's name is Brauer not Bauer, two R's in Brauer.
Furthermore, you present a preparation of CCl4 from S2Cl2 and CS2. When I posted about preparation(s) of CCl4 you said it is a carcinogen and
blathered about unsuitability for home chemistry.
Well, I could state similar opinions about questionable suitability of SO3 in home labs, if I wanted to, but, I don't happen to hold that opinion.
Whereas you seem to take the "Do as I say, don't so as I do" position. And this is why I have been compelled to wonder whether or not you are a
hypocrite, previously.
That is, you are perfectly content to conduct risky procedures at home, setting an example for others to follow. But when anyone else does the same,
you get all shrill and hysterical.
Double standard.
What would happen if that flask of refluxing SO3 and S2Cl2 shattered? Do you have any provision for containment? I guess that might have interfered
with yoiur photo shoot. Well, I would prefer to see appropriate safety procedures documented and illustrated along with the glassware.
As Dr Who once said (though it was probably Douglas Adams putting words in his mouth) "We scientists pay for our privilege to experiment at the price
of TOTAL RESPONSIBILITY."
SO3 is going to get someone hurt, someone who isn't properly trained or equipped, someone hapless. And then who will be responsible?
You will be. I suggest you ponder that.
Note that Brauer presented an alternative SOCl2 prep, reaction of PCl5 and SO2. This has similarities to the Kyrides method, in sense that SO2 is
treated with a powerful chlorinating agent.
But it suffers from the near unavailability of PCl5.
Another related method is the reaction of PCl3 and SO2Cl2. These are recirculated through a fixed bed (i.e., a cooled tube) of granular activated
carbon, and the result is a mixture of SOCl2 and POCl3 which can be readily fractionated.
Again the problem devolves to obtaining or making PCl3. In UK you could buy red P, and chlorinate it to PCl5, but not PCl3.
SO2Cl2 is easy, but nasty. It can also be prepared from sulfur chlorides by oxidation with Hg nitrate IIRC. Not really an attractive route.
Anyway I have come to regard SOCl2 as a very overrated reagent, one that can be replaced with half a dozen or so alternatives to advantage. I have
written about these alternatives extensivey and need not recite them again here, suffice it to say, thionyl chloride is passe.
[Edited on 19-4-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thank you Polverone, and thanks for the forum where I learnt about a lot of interesting stuff and gave me the opportunity to enjoy science. I thought
I'd take this chance to ask something I wanted to for a while - is it possible to enable editing for longer than 24hrs in the prepublication section -
or does the software not allow such differentiation?
@Sauron - I have no hard feelings - peace. I did not mean to slight your CCl4 thread in anyway, and appologise if a misunderstanding occured. Yes I
would feel very bad if what you say happens to someone with SO3. Every few lines I write about the danger of this compound. Surely no one can
misunderstand that. The method for making SO3 is in many textbooks - which however do not warn the reader nearly to the extent that I do. So another
way to think of this is that the graphic picture of what it does to polyethylene tubing will actual save people from accidents rather than enhance
them.
I cant agree with you that SOCl2 is passe. Not from where I am sitting. PCl3 is only available via P - which is much harder to make than the 3 steps
I describe (the whole operation, strating from S, Cl2 and NaHSO4, making the SO3 while the S chlorinates, takes about one working day). The you still
have to chlorinate the P - and it makes all sorts of hard to get rid of by-products to-boot! The other alternative you suggest - Kyrides reaction -
is good, if it works, and if you have the BzCl3. Most people I hazard to say dont, while chlorinating toluene that final one chlorine is a long haul
- I have done it, but it takes a whole day just for the chlorination. Then another day to do Kyrides reaction.
However I think his reaction is sweet in its own right!
[Edited on 19-4-2008 by len1]
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Len1, I have no desire to quarrel with you, either. I like your illustrated practical guides. I had no problem with your Cl2 thread, or your toluene
chlorination thread. And I do recognize that a properly equipped and protected amateur in a "home lab" setting can make, and make use of, SO3 with if
not exactly safety, at least with acceptable risk. I do rather wonder how many of the members, not to mention lurkers, know what proper equipment,
proper protection and peoper training mean in the context of SO3 and oleum.
A few years ago I wanted SOCl2 and could not buy it. Unimportable here. So I did the same research you did and I read the patents and Brauer and so I
bought 2 liters of 65% oleum at HUGE expense from Merck. That 4 Kg of oleum is still unopened. This isbecause in meantime I found I could do without
SOCl2. I am saving the soleum for something better.
Necessary equipment: hood. Shields. Containment trays.
Necessary protection: Full face shield, neoprene gloves, tygon labcoat.
And if I had no formal training in wet lab, and I mean beyond two years of instructional lab, I would hesitate to work on something as utterly
merciless and unforgiving as neat SO3.
I look around and I see members distilling nitric acid in their kitchens. I think that is just plain foolish. At the minimum that ought to be in a
hood, with some sort of cylinder or jar under and around the pot and receiver just in case. Anticipating potential accidents help avoid them turning
into disasters, as I'm sure you agree.
I'm no worry-wart. But I'm an old chemist, and as they say there are no old, bold chemists.
Suppose you are stirring a pot magnetically and a fuse blows or a breaker goes and the stirrer loses power. 5 times out of 10 the spin bar will not
slow down peacefully, it will at a certain point go crazy and sometimes knock the bottom out of the flask. Likewise, shaft stirrers if there is no
stop clamped onto them can come loose from the chuck and smack into the flask bottom with same result. Two of my least favorite scenarios, those. So I
spend time and brain cells preventing them.
Someone with less experience might not and might live to regret it.
Anyway thank you for your apology, I accept it entirely and also wish peavce and camraderie.
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Once again Len, Bravo! Very impressive work! I just love to read your very detailed reports, thank you for sharing all this hard work.
It appears you synthesis of S2Cl2 looks much cleaner and quicker than my attempt! If i ever try this again, i will follow your directions. Indded,
this is some strange concidence we started working on the same compound at the same time, and used it for completly different uses, without consulting
each other!
You have suceeded in one of the long-going dreams of the amateur science community, which is synthesis of thionyl chloride from available chemicals.
You prove that with enough determination, alot can be achieved.
Looking forward to your next ground-breaking report, whatever it will be 
|
|
|
Fleaker
International Hazard
Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
As usual, excellent work Len. I don't know how you find the time to do this--you must live by the creed ''action speaks louder than words''. I recall
my friend and lab partner did something in the same vein as you have albeit quite a bit ago--he mentioned it was quite possible to make SOCl2 although
I don't think he used straight SO3, but rather went the safer route with an oleum... He never posted it up here, and he did this before I met him
(among other experiments that I never got to see!). NERV did a good bit of work with these sulfur halides.
I must agree with Sauron though--stay away from straight SO3. If it can be avoided in a procedure, avoid it! You have seen yourself what I, Stefan,
and others have with regards to its reactivity. Oleums are nasty enough and I dislike working with them!
Nice to see this work, SOCl2 is useful and buying it is expensive, particularly the shipping!
Might you consider synthing chlorosulfonic acid?
Neither flask nor beaker.
"Kid, you don't even know just what you don't know. "
--The Dark Lord Sauron
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thanks Klute - you also do great work, and your write-ups are a pleasure to read. Indeed I had just come back from a long night at the lab doing the
stincking sulphur chlorination to see your report! The same happened before with benzaldehyde - Magpie was doing the synthesis at the same time. I
just used ordinary Australian garden sulphur, at $9 a 3kg bag. From what I got I suggest its 99% pure! Who needs AR sulphur from Aldrich or Merck,
when we spread higher purity stuff on the ground here.
I still am not happy with the result while the IR spectrum is not a perfect match. So I guess the next step is quinoline synthesis, as I cant buy it
anywhere - Skraups synthesis looks a beauty in its own right!
@Fleaker - thanks, I get the time very simply - by not sleeping. When I start breaking glass - I know its time to go home. While I agree with Sauron
100% that SO3 is a potential disaster - its also one of the most pleasant and informational experiments Ive done - no fuss no mess, just sit back and
enjoy. I would have hated to miss out on the experience (thanks go to garage chemist who also did this). Having said that I dont intend working with
SO3 every day of my life. Thats the beauty of amateur work - had enough of one reaction - go do something else. Its not like work, where you have to
repeat the same stuff over and over.
I did consider chlorosulphonic - its anhydride is generated - inter alia - in this experiment. But I find chlorosulphonation a yawn. Is there
anything interesting one can make with it? Len
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Very nice work Len! I agree that you have just conquered one of home chemistry's near holy grails. Many have talked about doing this - only you have
done it. There's a big difference IMO.
This is not in anyway meant to denigrate the warnings of Sauron. Our need as authors to do all that we can to warn people prior to attempting
potentially disastrous syntheses is a serious responsibility. I think you do a fine job of that.
| Quote: |
But I find chlorosulphonation a yawn. Is there anything interesting one can make with it? Len
|
I will just offer a couple of syntheses from my lab manuals. Perhaps they will whet your interest:
1. Preparation of sodium lauryl sulfate. Now you can make your own detergents! 
2. The sulfonation of acetanilide. Now you can make your own anti-biotics (sulfanilamide)!
And you thought chlorosulfonic acid was boring.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
May I propose a further Illustrated Guide topic to Len1?
The preparation of chlorosulphonic acid from anhydrous HCl and oleum (or SO3). Len has already done the hard part.
Like the dissolution of SO3 in oleum, this reaction is exothermic (highly so) and has to be done with rigorous exclusion of moisture (nothing new) and
these represent interesting challenges.
Obviously cooling with water would not ne a very good idea some some suitable coolant that would be inert to oleum, SO3, chlorosulphonic acid would be
advisable. Maybe a silicone? Just a suggestion.
This is a serious suggestion, no mockery involved. Chlorosulphonic acid is a sine qua non for sulphonations in many instances (at least, so get to the
sulphonyl chloride.) And where I am at least, it's a no-no, for the silly reason that it used to be used in military smokes.
I am perfectly content to see someone prudent, competent and properly equipped do a demanding and hazardous procedure safely. I discuss and propose
quite a few such myself, and will again. I just don't think they are every amateur's cup of cha. Nor does Len1, I bet.
Keep up the good work, Len1.
PS after posting this I saw that this idea has already been discussed. Sorry. But it was a logical extension.
[Edited on 20-4-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
BTW Len, is that glass wool your using to insulate your distn head? I've tried using plumbing insulation, but it's not pratical to fit around the
setup...
And how much TCCA did you use for your first chlorination?
I like your idea of using the U-tube to transfert the SO3, i find it pretty clever. Do you have schlenks? You could try collecting the SO3 in a small
schlenk, with a claisen to attach a condenser above it, then connecting it to your SCl2 still, and use a light flow of argon with a little heating of
the schlenk to transfert the SO3 in a graduall way. Using the U-tube in a similar way might eject big pieces of SO3 if it clogs up though.. Just a
idea.
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
| Quote: | Originally posted by len1
Thank you Polverone, and thanks for the forum where I learnt about a lot of interesting stuff and gave me the opportunity to enjoy science. I thought
I'd take this chance to ask something I wanted to for a while - is it possible to enable editing for longer than 24hrs in the prepublication section -
or does the software not allow such differentiation?
|
I have modified the software to allow 30 days of editing in Prepublication. Will that do?
PGP Key and corresponding e-mail address
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thank you very much Polverone - I think that gives plenty of time for improvement of articles and makes that section much better. I appreciate it
that you took the time to modify the software (I didnt really expect you would).
Klute, yes it is glass-wool, you can get bags of several cubic meters, and it stands up to 800C! Very useful stuff. I seem to remember European
hardware stores also carrying them.
The TCCA was 200gms, thats of course a bit excess - I just break up one pellet and put the lot in. Its enough for 2 moles of Cl2 and uses about two
250ml charges of 15% acid. Theres an error in the text (which due to Polverones kind modification of the forum) I can now change - 100gms S gave 208
gms of product.
Alas I dont have any schlenks. The first time I heard of them was in your posts. I might now acquire some. I am a physicist by profession (you I
presume are a chemist) so the only chem labs I worked in are ones attached to phys labs - and they never had such apparatus (or canulas for that
matter).
I was concerned about the U-tube ejecting a major part of the contents under pressure - as you say. I heated from the top, and waited till that
section melted before working further down, until all contents were molten and at bottom of U-tube. Thanks, I shall change the description as its
important for safety.
Sauron - I could make this acid - I dont know what Ill do with it though. I read somewhere that you can use it for chorinations, much as SOCl2, but
there were no details.
Thanks Magpie. I know chlorosulfonic is a useful chemical industrially but detergents are not that exciting. Antibiotics are more so, but I would
take internally only what my wife cooks up I suppose I could put them in a
petrie dish and see if they kill bacteria I suppose I could put them in a
petrie dish and see if they kill bacteria
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thank you very much Polverone - I think that gives plenty of time for improvement of articles and makes that section much better. I appreciate it
that you took the time to modify the software (I didnt really expect you would).
Klute, yes it is glass-wool, you can get bags of several cubic meters, and it stands up to 800C! Very useful stuff. I seem to remember European
hardware stores also carrying them.
The TCCA was 200gms, thats of course a bit excess - I just break up one pellet and put the lot in. Its enough for 2 moles of Cl2 and uses about two
250ml charges of 15% acid. Theres an error in the text (which due to Polverones kind modification of the forum) I can now change - 100gms S gave 208
gms of product.
Alas I dont have any schlenks. The first time I heard of them was in your posts. I might now acquire some. I am a physicist by profession (you I
presume are a chemist) so the only chem labs I worked in are ones attached to phys labs - and they never had such apparatus (or canulas for that
matter).
I was concerned about the U-tube ejecting a major part of the contents under pressure - as you say. I heated from the top, and waited till that
section melted before working further down, until all contents were molten and at bottom of U-tube. Thanks, I shall change the description as its
important for safety.
Sauron - I could make this acid - I dont know what Ill do with it though. I read somewhere that you can use it for chorinations, much as SOCl2, but
there were no details.
Thanks Magpie. I know chlorosulfonic is a useful chemical industrially but detergents are not that exciting. Antibiotics are more so, but I would
take internally only what my wife cooks up I suppose I could put them in a
petrie dish and see if they kill bacteria (but most stuff in the lab will do that anyway -so it will prove nothing)
[Edited on 21-4-2008 by len1]
[Edited on 21-4-2008 by len1]
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
I notice that you use what looks like a long Graham condenser for refluxing. Is that by intention or is that just what you have on hand?
I have a shorter Graham but it does not have ground glass fittings, so have never used it - it's an antique, given to me about 40 years ago. My West
condenser (similar to a Liebig) has the gg fittings so that's what gets used.
I don't even have a U-tube. You have indeed put it to some interesting uses.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Here its the technological nineteenth century - I have to use what I have at hand - any working improvisation will do. Im sure a West will work fine,
I have only a Leibig.
I have just found out that I cant get aniline unless I want it for work. So to get to quinoline, to purify the SOCl2 its going to be a long haul from
sodium benzoate, potassium nitrate, tin, and glycerine.
The U-tube is a relic from the SO3 experiment, where its perfect for batches up to 100grams in ensuring perfect condensation in the receiver and not
the condenser. If a flask is used in its stead, some condensation will occur in the condenser (because of SO3 vapour flux directly through the
flask), and then you have to get it out of there by passing hot water through the coils - something I wanted to avoid. You could also transfer the
SO3 from the U-tube to the flask - but again the less operations with SO3 the better!
[Edited on 21-4-2008 by len1]
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Is that in fact a Grahan or is it a spiral reflux condenser? A Graham really is not so appropriate for reflux because of the narrow bore. A spiral
reflux condenser is great, particularly the double walled sort. Lots of condensing surface and therefore capacity.
Sic gorgeamus a los subjectatus nunc.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Good point Sauron. It does indeed look like the cooling water goes through the coil.
Below is a picture of my Graham where the cooling water is in the jacket instead. That's the box in which I received it, mislabeled as "CONDENSER,
Liebig's." The distributor is Central Scientific Co (CENCO) which in the old days seemed to be the mother of all scientific equipment.

|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Hah. I did some business with Cenco. They were based in Houston as I recall. Shipped chemicals to me at age 16 or so, by truck freight, I had to go to
Red Ball or something like that to fetch them. I forget what I bought. More than 40 years ago.
Sic gorgeamus a los subjectatus nunc.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
As far as Im aware theyre both called a Graham condenser - here the second type with the water in the jacket is also called an inland revenue
condenser, and is good for hard-to-condense gasses such as ether (where I used it). It would be completely inappapropriate, even dangerous as a
reflux!
I do not have a West. When such i required I use a Leibig.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Graham condensers have a very narrow bore and are usually used only for vacuum distillations where the vapor path is through the narrow-bore (5mm or
so) spiral. They have a pressure drop across the spiral.
The condenser in which vapor passes through the outer portion and coolant through spiral isn't a Graham, it's just a spiral condenser, and effectively
it's a shortened, but very efficient, reverse of a Liebig. It is also possible to have an outer jacket with coolant, further increasing the cooling
capacity.
The West is same as Liebig except that the jacket is close to the core diameter so coolant velocity is increased for any given rate of flow.
The Graham would be utterly inappropriate in any application where vapor can solidify. Liquid condenses in lower part of spiral if used diagonallu and
has to be pushed through, causing back pressure. So for delivery the true Graham is best used vertically.
Wide bore Grahams are possible. But I have never seen one., The only condensers I have ever seen with wider bore coils are Buchi multi-cpol rotavap
condensers, and the coolant flows through the coils. The desktop ones have two coils, and the 20 L and larger models have at least 3 coils, and the
option of using two such condensers in series (Buchi E-type setup) for maximum condensing rates. I own a 20 L Buchi R152 with such a setup.
I would have thought SO3 would have been a no-go with a Graham, unless of course you kept coolant above the mp.
But obviously, you are succeeding, and therefore, all I am doing is kibbitzing. I really only meant to clarify Magpie's remarks.
Sic gorgeamus a los subjectatus nunc.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
| Quote: | Graham condenser
A Graham condenser has a spiral coil running the length of the condenser. There are two possible configurations for a Graham condenser. In the first,
the spiral contains the coolant, and the condensation takes place on the outside of the spiral. This configuration maximizes flow capacity since
vapors can flow over and around the spiral.
In the second configuration, the jacket tube contains the coolant, and the condensation takes place inside the spiral. This configuration maximizes
collected condensate, since all the vapors must flow through the entire length of the spiral, thus having prolonged contact with the coolant.
|
|
|
|
basstabone
Harmless
Posts: 24
Registered: 17-4-2008
Member Is Offline
Mood: No Mood
|
|
Do you have the patent for the purification? I made some quinoline the other day and can buy linseed oil from the hardware store.
|
|
|
| Pages:
1
2 |
|









