| Pages:
1
2 |
len1
National Hazard

Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Sulphur Trioxide and Oleum Using a Box Oven - Illustarted Practical Guide
This article is part of a series of Illustrated Key Syntheses in Chemistry which I intend to post, with the purposes and guidelines outlined in the
thread of the same name in the General Chemistry section. I shall attampt to ensure that all the relevant information is contained in a single post.
If sufficient reason arises for something to be changed or edited, I shall edit the entire post.
Aim
This article presents a simple procedure for obtaining oleum and SO3 from sodium bisulphate decomposition using a box oven capable of attaining 780C
maximum. The procedure uses efficient exhaust condensation, which enables the pyrolysis to be carried out in an ordinary fume-hood and at a stretch -
with no fume hood at all (not recommended in case of accidents). This condensation is of special importance as H2SO4 mists are notoriously difficult
to condense. A theory of the decomposition of NaHSO4 is presented, this is particularly relevant as it impacts directly on the importance of
carefully defining yield in this reaction, and explains yield loss.
Several threads on SO3 production, have already been posted - see for instance the excellent work of Stefan http://www.sciencemadness.org/talk/viewthread.php?tid=10217. I feel there is neveretheless a need for the present article for several reasons:
the present setup uses common materials and a lower temperature, sufficient substantial differences with previous work have been found (see findings),
an in-depth theory section which impacts experimental results is presented.
Findings
A 69% yield of SO3 based on available SO3 in NaHSO4 (assuming product is pure SO3). The main loss mechanism is evaporation of SO3 below 450C.
Oleum strength 90% as determined by distillation.
Oleum strength 102% +/-30% determined by titration.(The large error range is due to the fact that this calculation is very sensitive to
experimental error, see theory. Clearly a strength > 100% is impossible.)
An almost 100% H2SO4 yield based on NaHSO4 can be obtained.
End point temperature is a elatively low 780C due to uniform temperature distribution of box oven and use of retort, a geometry which enhances
evaporation of SO3/H2O from liquid reagent while minimising spattering.
Efficient vapour condensation system enables the pyrolysis to be carried out in an ordinary fumehood (the procedure can be conducted with no
fumehood - but this is not recommended should accidents arise). No SO2 evolution was detected.
NaHSO4 melts at 185-195, pure H2O evolution commences at about 220C, while at 260C white SO3 vapours (in the form of dilute H2SO4) start
evolving. The vapours become more concentrated so that at 340C conc. H2SO4 starts distilling through the viscous reagent. If the temperature is
raised 185C-450C in 5 hours the reagent never solidifies until the 'nominal' decomposition point of Na2S2O7 is reached.
It is thus seen that the usual description of the decomposition as a two stage processes 2NaHSO4 -> Na2S2O7 -> Na2SO4 the first releasing
H2O, the second SO3 is very much an idealisation. The processes are so intermixed, that the reagent is better regarded as a mixture of
Na2S2O7/Na2SO4/H2O or equivalently Na2SO4/SO3/H2O with the ratios varying with temperature. It appears very likely that the ratios also depend on the
temperature-time regime, which, if it were the case, would open a mechanism to increase yield. This was not investigated here, a single 5hr 185C-450C
temperature-time regime being used.
SO3 possesses some remarkable properties: traces of its vapour above the solid at 10C, invisible in a tube shielded from the atmosphere, fume
profusely when the insides of the tube are exposed to the open air (see pictures). Its vapour even at 10C instantly carbonizes anything organic - the
inside of polyethelene tubing is immediately carbonized (see pictures below), while black spots appear on any glassware containing even traces of
organic matter - this despite the glass having been washed in a dishwasher - it thus acts as a 'fingerprint developper'. If discolouration of the
snow-white SO3 is to be avoided glassware should be washed in piranha. Glass wool was found to easily withstand 800C, however its yellow pigment was
instantly carbonized - such wool should be heat-treated at 500C to burn-off the pigment.
It was found that Fe is not only strongly attacked by the SO3/H2O system at ~400C, but that it serves as a reaction vessel for its reduction
with almost 100% efficiency - no SO3 was seen to exit such tubes, however the smell of SO2 was marked. Investigation of the contents after cooling
revealed predominantly FeSO4, as well as FeS evidenced by evolution of H2S gas when water was added to the acid mix inside the tube. The extent of
the reduction of sulphur, from +6 SO3 to -2 in FeS, shows that the environment inside such tubes is so heavily reducing, that unless coated with a
melt insoluble salt, they are unlikely to be useful as reaction vessels in NaHSO4 pyrolysis.
Unlike SO2, SO3 possesses no warning smell, however its vapours are extremely corrosive and carcinogenic. They are fortunately visible - and
this should be used as a cue to avoid exposure.
Theory
The SO3 / H2O system
One way to regard the sulphuric acid system is as a mixture of the components H2O and SO3
a H2SO4 + (b - a) H2O == a SO3 + b H2O.
Note that this is not a reaction equation. The precise structure of such a system, in most situations is much more complex than either left or
right, rather its two alternative useful descriptions of the system. Mixtures where b >> a are called dilute sulphuric acid, b = a is
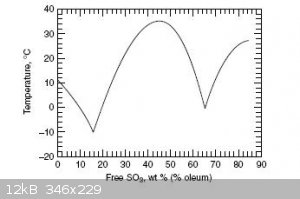
refered to as 100% H2SO4, while b < a is called oleum, both a fuming liquid and solid depending on its composition. This property of the b < a
oleum system is due to a peculiar non-monotonic phase diagram, which shows two maxima. Thus oleum is a solid at room temperature (20C) in the
interval 30%-60% SO3, and again at >75% SO3. A consequence of this peculiarity is that one can not determine whether one has >75% purity SO3,
or a lower concentration oleum by the appearance of a solid phase alone. A quantitative analysis needs be conducted. Due to the hysteresis in the
system a melting point determination is very inaccurate, rather a titration of the homogeneous product, or evaporation of SO3, is required.
Returning to the equation above, b = 0, corresponds to pure SO3, a white deliquescent fuming solid with a melting point of 16C and boiling point of
44C. This very narrow range of temperatures where SO3 is a liquid, is the source of its condensation problems during its production by pyrolysis.
SO3 easily polymerises (enhanced by H2O, inhibited by P2O5), with three modifications, with melting points 32C, 62C, and 95C, respectively.
If one tries distilling sulphuric acid with b >> a, initially a dilute acid distils, with the fraction of the SO3 progressively increasing until
the maximum boiling point at 337C of the liquid/vapour azeotrope of 98%/2% H2SO4/H2O is reached. After this no further concentration in the SO3
component can be obtained. Hence distilation can not be used to prepare oleum/SO3, it is also quite wasteful for preparing conc. H2SO4 from the weak
acid.
The SO3 / H2O / Na2SO4 system
A way to obtain oleum is provided by the acid salt NaHSO4. One way to regard the anhydrous salt is as an equimolar mixture of 100% H2SO4 and the
basic salt Na2SO4 (it is basic being the salt of the weak acid NaHSO4). Certainly an aqueous solution of NaHSO4 is perfectly equivalent to such a
mixture
2NaHSO4 == H2SO4 + Na2SO4
Again this is not a reaction, but an equivalence in the sense of the first equation. Since NaHSO4 has a relatively high pH (1mol/L has pH~2)
we see from the equivalence above that Na2SO4 has a marked effect on the pH of H2SO4 solutions with an equimolar mixture raising the pH of 1mol/L
H2SO4 from pH << 0 to pH ~ 2 (buffering). Na2SO4 thus being a weakly basic salt explains why the pyrosulphate can be decomposed at
fairly low temperatures in what follows (compare to CaSO4 -> CaO + SO3 at 950C < T < 1200C, CaO being a much stronger base).
The formula above, which was presented as being merely an equivalent representation, turns out to be a realisable reaction in practice. Indeed H2SO4
can be distilled from the acid salt NaHSO4. Here we have found the distillation is 97% efficient. Collecting the distillate as a single fraction
gives 100% H2SO4 in 97% yield. However, unlike the SO3/H2O system, in the Na2SO4/SO3 system, introducing a cut in the distillate fraction will yield
us oleum, with the lower b.p. fraction being therefore a correspondingly weaker acid (since a combination of the fractions must give 100% H2SO4).
We can say that the addition of Na2SO4 to H2SO4 breaks the azeotrope.
This distillation is generally 'idealised' as two reactions the first being evaporation of H2O
2NaHSO4 -> Na2S2O7 + H2O 200C < T < 650C
the second being evaporation of SO3 leaving behind a weakly basic salt
Na2S2O7 -> Na2SO4 + SO3 260C < T < 780C
The temperatures quoted are the ones I have measured here rather than those in the literature, which suggest almost no overlap of the reactions. In
actual fact both reactions occur at the same time over a wide temperature range and the system consists of a complex mixture of NaHSO3/Na2S2O7/Na2SO4
at all but the bottom and top temperature ranges. Nevertheless substantial H2O evolution occurs at 200-260C while almost no SO3 is being evolved, and
substantial SO3 doesnt get released until 780C, when no water is essentially left behind, and this explains the separating power of the NaHSO4
distillation.
Nonetheless substantial SO3 is released below the 'nominal' decomposition temperature of the pyrosulphate at 450C, about 30% in the present
experiment, which indeed is the major component of the yield loss in the 69% SO3 yield quoted here. In addition some H2O is released with the SO3 at
the low-end of the SO3 evolution range of 650C. This demonstrates that the pyrosulphate is a convenient idealisation for what in reality are complex
mixtures of Na2SO4/H2O/SO3 existing at the various temperatures.
The efficiency with which the various components H2O/H2SO4/oleum/SO3 are separated is dependent on the homogeneity of the NaHSO4 composition
(distilation of 100% H2SO4 added equimolarly to Na2SO4 without intimate mixing will just return the 100% acid) and the time - temperature regime.
Definition of yield and strength of oleum
One has to define carefully what one means by yield in such a system. The simplest definition is percentage yield H2SO4 based on sulphate in the
NaHSO4 - with 100% yield corresponding to distillation of 50% of the total sulphate - with Na2SO4 forming the undistilled remainder. However
generally one is interested in obtaining oleum. The yield for such a procedure, which requires one to take a cut on the distilation can be defined as
so much percentage yield for oleum of quoted strength. Obviously the higher the strength of oleum desired, the higher the temperature at which the
product starts to be collected, and the lower the yield.
Generally one defines the strength of oleum as a percentage of the weight of free SO3 to total weight of oleum. Free SO3 is defined as the weight of
SO3 after the amount of SO3 bound as 100% H2SO4 has been subtracted out. In practice, for an SO3/H2O system with weight composition a SO3 + b H2O
this prescription amounts to performing the arithmetic:
[a - b * (80/18)] / (a + b)
100% SO3 (b=0) then corresponds to 100% oleum, while 100% H2SO4 is zero percent oleum - with no free SO3 being able to be distilled off.
Within this formula lies a difficulty in determining the strength of oleum with any precision experimentally (as will be seen below). Because of the
small molar mass of H2O its weight is multipluied by a factor of 4.4 in the formula, which, on performing the substraction, has the effect that small
errors in H2O weight lead to large errors in oleum strength. Put another way, it only takes 18 grams of water to convert 80 grams of SO3 (100% oleum
- a fuming solid evaporating at 44C with all SO3 available), into ordinary sulphuric acid boiling at around 300C (0% oleum with no free SO3
available). Hence glassware in which SO3 is generated and reacted should be oven dried to avoid loss of yield.
In the present article a 460C cut on the distilled fraction gave oleum of strength 80-100% (the large uncertainty for the reason explained above) with
69% yield based on free SO3 determined by titration, while the percentage yield H2SO4, based on total fraction with no cut-off temperature was 97%.
Acid-base sulphate and bisulphate equilibria
pH of HSO4- solutions
The bisulphate anion acts as a weak acid due to the low value of the following equilibrium constant
HSO4- -> SO4-- + H+, [SO4][H]/[HSO4] ~ 10^-2 -> pKa ~ 2
pKa = 2 is sufficienly large compared to pK = 7 in water that we may neglect protons contributed by water. Its also sufficiently small that in
solutions of order 1 mol/L loss of HSO4- to dissociation can be negleted. If c is the latter concentration
[H] = sqrt(c 10^-2) -> pH = 1 - 1/2 log c
pH of SO4-- solutions
These solutions are weakly basic, and we have to use the proton equilibrium in water
[H][OH] ~ 10^-14
and the mass balance equation for the protons [H]
[OH] = [H] + [HSO4]
We neglect SO4 dissociation, whose concetration is c, again due to the small pKa above, hence
[H]^2 + [H][HSO4] = [H]^2 + [H]^2 c / 10^-2 = 10^-14
It follows
pH = 8 + 0.5 log c
A 1 mol/L Na2SO4 solution is expected to have pH ~ 8.
Method
All operations described below were carried out in a fume hood, wearing a gas mask, latex gloves, and with a
drought established in the laboratory.
Ambient temperature in the lab 25C.
Although the NaHSO4 distillation can be carried out in a single step, taking a cut at the desired temperature, I did not wish to expose myself to
excess SO3 fumes such as entailed in making connections when replacing receiver and condenser. Hence the process was carried out in two steps with
the lower temperature part taking place outside in a shed.
There is a precedent for this in a procedure described in the Practicum on Inorganic Chemistry, Editor Yu. Tretyakov, p. 235
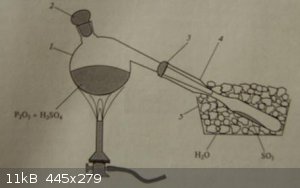
| Quote: | | After the completion of SO2 evolution by the action of sulphuric acid on sodium sulphite, the contents are placed in a porcelain crucible on a sand
bath and heated till they solidify. The solid contents are ground in a mortar and placed in a high-T retort, whose outlet is placed in iced water.
The retort is heated with a propane flame, condensing the white SO3 vapour inside a test-tube, which is subsequently sealed. |
Step 1 - Distillation of low temperature fraction
SO3 released by the build up of pressure inside a warming container and sprayed in the eye will blind you
instantly. Please wear glasses and be careful.
329 grams of NaHSO4 (pH-down for pools) were placed in a porcelain crucible and heated on a shielded sand bath. The exhaust fumes were not collected,
so the procedure was carried out in a shed. The temperature of the bath was initially held at 250C until the salt melted - this occurred at about
190C as measured at the melt surface by an IR thermometer. When the melt reached 210-220C water vapour evolution commenced - but ceased in about
20mins with the melt at 220C. A picture of the melt at this stage is shown below.

The temperature of the bath was now raised to 350C - when the melt reached about 260C sulphuric acid vapour evolution commenced - as evidenced by a
deep white smoke. Vapour evolution continued with the bath temperature rising to 285C in about an 90mins, the temperature being limited by the heat
carried away by the evaporating H2SO4. At this time the bath temperature was set to 450C, whereupon the surface temperature rose to 337C where is
stayed relatively constant for about an hour - due presumably to evaporation of the H2SO4 azeotrope. Distillation was stopped when the melt surface
temperature reached 425C (about 450C in bulk), at which stage the reagent was fuming and had the form of a viscous liquid impregnated with tiny
SO3/H2O bubbles. At no stage, from the nominal melting point of the NaHSO4 to the nominal melting point of the Na2S2O7 did the melt solidify. It is
obvious, that quite a substantial amount (as turned out subsequently almost the entire yield loss) of SO3 was evaporated at this stage. It is
possible that some of this could be avoided by shielding the melt from the atmosphere - as reaction of SO3 vapour with atmospheric moisture (in
addition to that evaporating off the surface of the liquid), SO3 + H2O -> H2SO,4 decreases its partial pressure substantially and enhances
evolution of further SO3 by Le Chattliers principle. It is also probable that yield could be enhanced by maintaining the melt in the 240-260C range
for longer than the 2hrs here. However evaporation gets progressively slower, and as NaHSO4 is a very cheap reagent, experimental time was considered
the more valuble comodity.
The remaining material now weighed 272 grams. The corresponding weight for a pure Na2S2O7 composition is 304 grams. Hence if one assumes the
material consists of a Na2S2O7/Na2SO4 mixture (all H2O evaporated) 32 grams SO3, or 29% of total available SO3, co-evaporated with the H2O as H2SO4 in
the previous time-temperature regime.
The sulphates shrink more than porcelain on cooling (as opposed to an NaCl/CaCl2 mix which cracks porcelain), and hence are easily separated by
inversion and light tapping. The material was then broken up into small lumps and powder in a mortar as seen below.

Step 1 - Pyrolysis and high temperature cut
The material is now placed in our reaction vessel, which is a 50cent vinegar dispenser from Cheap-as-Chips. This container will easily withstand the
800C or so encountered in this experiment, as it was fired in a kiln at almost twice that temperature. It is however meant to operate at temperature
gradients of the type encountered in everyday life, and hence will crack if its oulet is at temperatures of 600C or so below oven temperature, such as
would occur in the present experiment. Hence, a piece of borosilicate tubing - which has one of the lowest expansion coefficients of any glass and a
softening point of 821C - was designated to take this temperature gradient, and was fitted snugly over its outlet, with a glass-wool filler sealed by
twisting in between. If one wished to eliminate carbon impurities due to subsequent carbonization of the pigment if the wool is coloured, the pigment
should be burned-off at 500C. 97 grams of the mixture from the first step were placed in the reagent vessel which is seen below. Due to its retort
shape bumping should be minimised, while tilting gives the melt maximum exposed surface. Despite the simplicity of the arrangement it will be seen
subsequently that this step gives almost 100% yield.

Next a 120 degree bend was inserted into the quickfit end of the tubing. Since the temperature of this joint reaches 200C degrees, and I did not wish
the joint to freeze - while grease can not be used with SO3 - I inserted some glass wool into the joint. This arrangement worked well - with the
exception that the small amount of pigment in the wool was carbonized and added a tint of brown to the first oleum/SO3 distillate. A long tube with
quickfit ends, to minimise losses, was used as the main (air condenser) leading to a U-tube maintained in water cooled below 10C, where the product
collects. Since H2SO4 evaporation is notorious for generating hard to condense fogs, a reflux condenser was fitted to the other end of the U-tube,
and cooled by a small water pump from the cooling container. The exhaust from the reflux condenser was vented through polyethelene tubing -
this could be passed outside - but as it turned out, the cooling was so effective that it produced no fumes. Since a box oven rather than a crucible
oven is used the oven door was removed and the exit sealed with an asbestos plank and porcelain plate cut to enable the borosilicate tube to pass. The
entire arrangement is seen below.

The oven temperature was initially set to 300C and upon equilibration, raised about 200C every hour. Up to about 520C nothing could be seen, except
1-2 H2SO4 drops condensing and dripping down again in the lower half of the 120C bend - this thus formed a kind of fractional distillation apparatus.
The same mixture at stage 1 produced a great deal of smoke at 450C, hence this was mostly due to air moisture, which was absent inside the reactor
tube. At 550C a white fog filled the air condenser and intensified at 600C. The fog extended to the U-tube, but the reflux condenser remained clear,
showing the effectiveness of sulphuric acid condensation. Effectively no product was collected in this temperature range. At 650C the insides of the
U-tube frosted over with oleum snow and some product was collected - this temperature was maintained until the air condenser was clear again.

Raising the temperature to 700C resulted in a mass flux of white smoke and liquid streaks down the air condenser, with oleum drops dripping down the
120C bend. The smoke worked its way up the glass wool and carbonized the pigment. The U-tube was now completely coated with solid - the smoke
extended to the first few coils of the condenser - but no further. The top of the condenser was clear. When the air condenser was clear again, the
temperature was raised and the procedure repeated at 750C and 780C - the latter producing the last large efflux of SO3. After this raising the
temperature to 800C and then 820C - considered the limit of this setup - produced no more efflux. Subsequent weighing in the next section shows
almost 100% distillation efficiency.
Temperatures measured with an IR thermometer showed a temperature of about 220C on the borosilicate glass tube at a point 2cm from where it exited the
oven, and 120C at the junction with the 120 degree bend, at the highest temperature setting of the oven - 820C. The air condenser heated to about
80-90 C when passing material.
The glass tube showed no-annealing indicating the temperature could have been taken higher if needed. Pictures of the oleum drops, streaks, and white
smoke are shown below. Note the almost complete carbonization of the first part of the polyethelene tubing on the efflux from the reflux condenser.
This was achieved with no visible fumes in the top half of the condenser at all - i.e. at the SO3 residual vapour pressure for 10C. In this way the
tubing serves as a very useful indicator of the progress of SO3 vapour out of the system. Note that the radiation emanating from the oven appears
violet (which would correspond to about 5000K) this is a frequently seen camera artefact - the actual colour at the maximum temperature was bright
orange.


The oven was turned off and the U-tube removed. The condenser - which was clear throught the procedure - started to smoke profusely when its insides
were exposed to air (see below).

The U-tube was disconnected and the contents weighted in at 26.9 grams. It was quickly wraped in cellophane and placed in a fridge. If this is not
done the stoppers are likely to pop-out due to build up of pressure as the SO3 warms. The fumes this releases are extensive and dangerous. I must
admit to being horrified at the danger this material obviously poses.

It follows that SO3 sprayed in the eye will blind instantly. Please wear glasses and be
careful
Calculations, yield, and oleum/SO3 stregth
The material left in the retort after distillation weighed 67.2 grams. The amount expected from the reaction
2NaHSO4 -> Na2SO4 + SO3 + H2O
with a fraction of 97/272 of the initial weight of 329 grams of NaHSO3 used in the pyrolysis is 69.4 grams, the distillation efficiency is thus within
experimental error 100% (the fact that its slightly over can be explained by the presence of moisture in the 'anhydrous' salt). This supports the
earlier finding that at 780C the reaction is essentially complete.
The 67.2 grams Na2SO4 together with the 26.9 grams of the SO3/H2O oleum distilled add up to 94.1 gram - thus giving a 97% yield in the pyrolysis -
this is the quoted yield on H2SO4 - essentially no SO3 or H2O was lost outside the system.
The available SO3 content of the initial 97 grams, corresponding to 97 * 329 / 272 = 117 grams starting NaHSO4 is 39.1 grams. If one assumes the
entire product gathered is pure SO3, the yield based on available SO3 in the initial NaHSO4 is 26.9 / 39.1 = 69%. However, as pointed out in the
theory section, the final product is actually oleum, whose strength is yet to be determined. The water presence in the latter will alter this yield
(only slightly because of the low molar mass of water).
The final step is to find the H2O/SO3 ratio in the product and determine oleum strength for our 450C cut. This can be done in two ways. The first is
to titrate the oleum. However this is very inconvenient as it exposes one to SO3 fumes. An alternative method is to use the fact that we have almost
100% distillation efficiency. This means the H2O/SO3 ratio of the reagent from step 1 is the same as of our product oleum - and so the reagent can be
titrated either with BaCl2 to determine sulphate content, or a base to determine SO3 content in the form of Na2S2O7 and NaHSO4. We use the latter
method.
11.2 grams of the product from step 1 were dissolved in about 100ml H2O and titrated against a 2% NaOH solution ([NaOH] known to an accuracy of 3%].
3.62 grams NaOH raised the pH from an initial pH < 1 to 8.40 at the end point. It is interesting that the pyrosulphate was self-indicating - so a
pH meter is not really needed. It changed from colourless to a deep yellow during the addition of the last 1/2 ml, and a small amount (about 0.1
grams) of a yellow-orange precipitate (presumably colloidal sulphur) formed.

This evidenced the presence of 3.62 grams SO3 in the 11.2 grams of step 1 product (coincidently NaOH neutralizes SO3 in a 1:1 weight ratio). The
Na2SO4 content in the latter can be determined as follows: since distilling 97 grams of product gives a residual of 67.2 grams, the Na2SO4 content of
11.2 grams of product is ~ 7.7 grams. The H2O content is then
11.2 (+/- 1%) - 3.62 (+/-3%) - 7.7 (+/-2%) = (-0.12 +/- 0.3)grams H2O
The amount of H2O in the 11.2 grams of product then ranges from 0 to 0.28 grams (obviously we have to exclude negative values). Plugging this into
the formula for oleum concentration gives oleum > 72%, but thats the lower 70% confidence limit.
Distillation of SO3 from Oleum
As mentioned in the introduction the amount of SO3 in oleum can be determined by evaporation. Since in this case we are directly determining the
weight of SO3 which can be distilled from a given strength oleum, rather than the weight of H2O, this method is much more sensitive than the titration
in the paragraph above. The distillation is hazardous, due to once again possible exposure to SO3 fumes, and this part must certainly must be
conducted in a fume-hood.
As with any distillation a cut must be chosen, and this was taken as 100C for convenience, meaning a water bath can be employed. Obviously the
temperature is a compromise between the amount of H2O coming over with the SO3 and the amount of free SO3 left undistilled. The water vapour pressure
above 0% oleum (100% H2SO4) at 100C is minimal, while the oleum remaining at that temperature is 37% free SO3, from the following table:
% Oleum b.p.
0% 290C
10% 175C
20% 140C
37% 100C
The optimum cut for many applications is actually between 140-175C being less wasteful, while still keeping the vapour pressure of H2O negligible.
The U-tube filled with oleum from the previous part was connected as shown in the figure below, with the tube being immersed completely in water
heated from below. Its initial temperature was about 30C, and a final temperature, established by adding boiling water and heating was 100C.

The oleum melted at about 55C bath temperature - it would indeed have been possible to pour it between the containers at that stage. This shows the
SO3 was not in one of the two higher m.p. modifications (59C and 95C), which can be attained by remelting and solidifying, and enhanced by the
presence of water. The SO3 started boiling at that temperature - and initially white fumes filled the chilled container subsequently being replaced
by a flux of clear drops - this always happens when SO3 passes into a container with a hint of moisture present, once that is combined, pure SO3 is
colourless. An air blower was used to warm the 90 degree still bend to ensure no condensation of SO3 there.
After as much SO3 as possible at 100C collected in the chilled flask, the still bend was disconnected and both the U-tube and flask stoppered. See
picture below.

The small amount of organics discolouring the SO3, mentioned above, stayed with the 37% oleum in the U-tube. The interchange of stoppers, performed
with the oleum at 100C is the only procedure to generate any SO3 smoke in the entire experiment. The bend smoked profusely, so the fume-hood could
not cope, the laboratory had to be briefly evacuated.
The weiht gain of the flask was 22.4grams, the weight of 37% oleum was 4.2grams, meaning a loss of only 0.2gram or <1% oleum in the distillation.
The 37% oleum contained 1.5gram SO3, with 2.7gram being 100% H2SO4. Together with the 22.4gram pure SO3 this means initial oleum strength prior to
the distillation was 90% - in agreement with the titration figure. The distillation of 90% oleum to essentially 100% oleum, or pure SO3, was 94%
efficient.
Being concerned to achieve maximum yield for this very simple distillation I waited until the last drops of SO3 had collected in the receiver (the
flux for the last few drops was low and took about 5mins). These promptly froze in the narrow tube creating an icicle, this could not be removed by
heating the upper part of the bend from the outside, part of it was shaken off when the bend was removed, nonetheless the remainder, while not
contributing to the yield, was clearly < 0.2grams. It had to be taken outside (see picture below) as it threatened to fill the whole lab with a
mist, and the procedure shows the importance of maintaining a reasonable flux of SO3 during the distillation and not cooling the flask too much.

Health effects
SO3 has no smell but fortunately minute amounts fume profusely in the atmosphere (it is used as a smoke agent in battle), and hence provide easy
warning. It is extremely agressive on metals and anything organic as can be seen by the instant carbonization of polyethelene piping by minute
amounts of SO3 due to its vapour pressure at T < 10C. It 'develops' as black dots the smallest traces of organics, such as left by the hands, on
all glassware, especially particles trapped in quickfit joints even after several washes. It is also carcinogenic - I provide the following quote as
a warning
| Quote: |
Strong inorganic acid mists containing sulfuric acid are known to be human carcinogens based on sufficient evidence of carcinogenicity from studies in
humans that indicate a causal relationship between exposure to strong inorganic acid mists containing sulfuric acid and human cancer. Occupational
exposures to strong inorganic acid mists containing sulfuric acid are specifically associated with laryngeal and lung cancer in humans. Steenland et
al. (1988) reported on studies of one U.S. cohort of male workers in pickling operations in the steel industry, which showed excesses of laryngeal
cancer (approximately two-fold) after adjusting for smoking and other potential confounding variables. In a ten-year follow-up, Steenland (1997)
reported a two-fold laryngeal cancer rate ratio consistent with previous findings from this cohort. |
Fortunately, if the procedure is carried out as described here, no exposure to detectable amounts of SO3 should occur.
Quote from http://ntp.niehs.nih.gov/ntp/roc/eleventh/profiles/s164sulf....
Conclusion
Oleum/SO3 of at least 72%, but more likely closer to 100% has been obtained, at 69% yield based on NaHSO4 in a two-step procedure. The procedure uses
apparatus easily accessible to the amateur, and can be carried out in the normal laboratory. SO3 is aggressive and will easily blind.
[Edited on 21-4-2008 by len1]
Edit by Texium: fixed broken image links
[Edited on 6-9-2023 by Texium]
|
|
|
ScienceGeek
Hazard to Others
Posts: 151
Registered: 22-1-2008
Location: Norway
Member Is Offline
|
|
Wow!! Once again len1: Beautiful work with both the experiment and writing! 
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thank you very much.
|
|
|
Pulverulescent
National Hazard
Posts: 793
Registered: 31-1-2008
Member Is Offline
Mood: Torn between two monikers ─ "hissingnoise" and the present incarnation!
|
|
Jeez! Yeah, I haven't even read the post (yet) for admiring the images!
I'd happily sacrifice some lesser-used digits for that kind of glassware.
I'm saving reading the post 'till later---oleum is one of SM's holy grails.
P
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Great work as usual len1.
It is obviously idealistic of me to assume that the product I obtained was pure SO3. A determination of purity of my product would be a good addition
to my procedure.
You should mention that a third good method of determining the purity of the obtained SO3 and therefore the yield is by redistillation of the product.
This can be carried out in a standard ground glass distillation setup, with no cooling water flowing through the condenser,
and allows for nearly complete separation of SO3 from 100% H2SO4 due to the large difference in boiling points.
This way, the SO3 is also obtained in a pure state as the distillate and can be weighed directly.
Your usage of a vinegar dispenser is creative and cost-efficient.
This is made of some kind of porcelain, right?
I have never seen a similar thing where I live.
I would definately be happy about a retort with a larger capacity than 100g NaHSO4, but this would obviously also require construction of a suitable
larger furnace.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thanks Garage Chemist and Pulverescent.
I did consider distilation of the SO3 but the b.p. of oleum rises with decreasing SO3 content to about 290C at 0% oleum, while the water vapor content
of the distilling mixture also rises, so the separation wouldnt be perfect - though granted much more sensitive than the titration method I use. To
be honest I was quite shocked when I saw what SO3 vapour was doing to organic material and so chose the simpler but more inaccurate route.
I know what you mean regarding the vinegar dispenser. Last year while on holiday I searched your neck of the woods for suitable porcelain dishes when
I was still hoping to make Na by NaCl electrolysis and was surprised to find how poor the cereamics selection is. We have much greater variety here
due to most of our products being made in China.
|
|
|
Pulverulescent
National Hazard
Posts: 793
Registered: 31-1-2008
Member Is Offline
Mood: Torn between two monikers ─ "hissingnoise" and the present incarnation!
|
|
Garage chemist, your procedure is seeeriously interesting, too; I 'must get more glass!
It's manana with me, though!---grr--this procrastination!
I'm not discarding my ancient idea of trying to electrolyse H2S04 to remove H20 to the point where oleum is produced, though.
Well, not unless someone's found insurmountable obstacles to that process!
And I have an inert anode!
P
|
|
|
MagicJigPipe
International Hazard
Posts: 1554
Registered: 19-9-2007
Location: USA
Member Is Offline
Mood: Suspicious
|
|
Surely that wouldn't produce oleum but (in theory) just make 100% H2SO4. Something tells me it's not that easy though.
Has anyone actually tried that?
"There must be no barriers to freedom of inquiry ... There is no place for dogma in science. The scientist is free, and must be free to ask any
question, to doubt any assertion, to seek for any evidence, to correct any errors. ... We know that the only way to avoid error is to detect it and
that the only way to detect it is to be free to inquire. And we know that as long as men are free to ask what they must, free to say what they think,
free to think what they will, freedom can never be lost, and science can never regress." -J. Robert Oppenheimer
|
|
|
Pulverulescent
National Hazard
Posts: 793
Registered: 31-1-2008
Member Is Offline
Mood: Torn between two monikers ─ "hissingnoise" and the present incarnation!
|
|
Yes, AFAIK, 100% H2S04 is a non-electrolyte, not being ionic, but back in the day that didn't occur to me.
Getting it to that concentration, electrolytically, probably would be very difficult.
But H2S208 is essentially oxidised oleum!
'Tiny chink of light at the end. . . etc..
P
|
|
|
microcosmicus
Hazard to Others
Posts: 287
Registered: 31-12-2007
Member Is Offline
Mood: spin up
|
|
No, 100% H2SO4 dissociates to H3SO4+ and HSO4- with an
equilibirium constant of 2E-4, as compared to the dinky 1E-14
for H20 dissociating to H30+ and HO-, so it is a far better
electrolyte by many orders of magnitude. for instance, the other
week, I electroplated Cu out of a solution of CuSO4 in 93% H2SO4,
so there is no problem doing electrolysis using sulphuric acid as
a solvent.
|
|
|
Pulverulescent
National Hazard
Posts: 793
Registered: 31-1-2008
Member Is Offline
Mood: Torn between two monikers ─ "hissingnoise" and the present incarnation!
|
|
Many thanks for that, microcosmicus! 'Shows how little I know.
Though we're deviating from the substance of the thread somewhat, what you say puts a different complexion on things (lights up the tunnel).
Are those ions formed in response to an applied potential, or are they normally present in 100% acid?
And did you use an inert anode in your electrolysis?
P
|
|
|
Pulverulescent
National Hazard
Posts: 793
Registered: 31-1-2008
Member Is Offline
Mood: Torn between two monikers ─ "hissingnoise" and the present incarnation!
|
|
'Sorry micro, even Faraday would have difficulty with the first question!
P
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Nice. So it is confirmed that the SO3 obtained from NaHSO4 was already close to being pure.
Do you have any plans on further syntheses or experiments with the SO3? Chlorosulfonic acid, DMS, DDT?
|
|
|
Saerynide
National Hazard
Posts: 954
Registered: 17-11-2003
Location: The Void
Member Is Offline
Mood: Ionic
|
|
Wow. Execellent write up and photos. That was awesome   (and that PE tube is scary) (and that PE tube is scary)
"Microsoft reserves the right at all times to monitor communications on the Service and disclose any information Microsoft deems necessary to...
satisfy any applicable law, regulation or legal process"
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thanks Stefan. I was considering DMSO as its useful, what can chlorosulfonic acid and DDT be used for?
Yes SO3 is quite charming as it quitely collects behind glass. I remember thinking 'what beautiful frosting' while looking at it crystalizing in the
U-tube, it was only half an hour later that I noticed a long section of previously clear PE tubing was completly charred. And then of course
disasembling the apparatus the entire hood was flooded with white smoke.
[Edited on 14-4-2008 by len1]
|
|
|
markgollum
Hazard to Self
Posts: 53
Registered: 21-2-2004
Member Is Offline
Mood: No Mood
|
|
SUPERB WORK!!
I am again impressed with the quality and usefulness of your practical guide.
If I had SO3 I would use it to make thionyl chloride, SOCl2. This is a very useful general reagent used to convert acids and alcohols to acid
chlorides and alkyl chlorides respectively.
ROH + SOCl2 -> RCl + SO2 + HCl
Same thing with acids, It even works with sulfonic acids.
The main advantage of thionyl chloride over PCl3 (which also serves to chlorinate acids and alcohols) is that you don't have to separate the
phosphorus-containing byproduct from your desired product as the SO2 and HCl just bubble out of the flask. I have seen SOCl2 used in inorganic
chemistry as well, but I can remember what it did.
To make it
SO3 + SCl2 -> SO2 + SOCl2.
I think it proceeds under mild conditions.
A post from Chris the Great halfway down this thread http://www.sciencemadness.org/talk/viewthread.php?tid=1439&a...
Indicates that the conditions are mild.
However, Sauron indicates that this reaction is a royal pain in the ass on the first post of this thread.
http://www.sciencemadness.org/talk/viewthread.php?tid=8311&a...
[Edited on by markgollum]
|
|
|
CyrusGrey
Hazard to Others
Posts: 123
Registered: 20-1-2007
Location: USA
Member Is Offline
Mood: Oooh! Shiny!
|
|
I have put this article on the HCS website with len1's permission. I just finished updating it to the newest version.
http://www.homechemistry.org/index.php?title=Sulphur_Trioxid...
Your welcome to use the in depth articles section of the HCS for other prepublication articles, len. It should be easier to edit it than repost the
article repeatedly. That leaves more room for discussion in the threads here.
|
|
|
ShadowWarrior4444
Hazard to Others
Posts: 226
Registered: 25-4-2008
Member Is Offline
Mood: Sunlight on a pure white wall.
|
|
Would it be possible to obtain more SO3 from the Bisulfate by simply placing a desiccant such as MgSO4 in line before the air condenser, and perhaps
skipping step one--directly inserting bisulfate into the furnace. Or will the SO3 rip the water of crystallization off most desiccants forming H2SO4?
(Perhaps P2O5 would be able to dehydrate the H2O/SO3 stream, but this is not particularly OTC.)
[Edited on 5-12-2008 by ShadowWarrior4444]
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
Nice job, my respect.
Okey this is just something that popped up in my head while reading this and it just might be something. Just a suggestion.
Maybe it is possible to use a microwave instead of an oven.
Super high temperatures can be reached.
Iam not sure about how this would alter the decomposition and thus the obtained product.
Also the fractional part could be difficult to control.
But still if there is any chance it might not be so bad.
I would be quick, dirty and OTC.
What a fine day for chemistry this is.
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
Oke a microwave oven is a bad idea.
I placed 0.5 gram of sodiumhydrogensulfate on a watchglas en placed it in a microwave oven at 100% power (800watts)
The timer was set to 30 seconds, nothing happend no melting or decomposition what so ever.
Another 30 seconds was run, after about 15 seconds an explosion occurred.
The watch glass was scattered into hundreds of pieces, the microwave survived.
Also there was a large could of vapour.
The vapour was probably consisting out of water (based on smell).
What I think happened was that there was water trapped in the cristals and in could escape causing super-heating and thus explosive ejection.
Another lesson learned today.
No one was hurt
A lot of substances dehydrate perfectly in a microwave, this one clearly not.
Even under adapted conditions i cant imagine how this could work.
I would have been nice to make pyrosulfate this way.
What a fine day for chemistry this is.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by User  |
What I think happened was that there was water trapped in the cristals and in could escape causing super-heating and thus explosive ejection.
|
No, it was just the solid that melted, that's all. The only problem is in that ionic melts have several magnitudes higher microwave absorption
compared to solids. When the MW source is 800W suddenly most of this energy is absorbed by the the small amount of the melt. This essentially means
lots of broken glass and the owen full of decomposition products.
It would be nice if you would read the theory about microwaves before engaging in potentially dangerous games with it. The least you could do was to
check the melting point of sodium bnsulfate (59°C monohydrate and 315°C anhydrous). You can not use such low melting materials in a MW oven without
power control, since even though their MW absorption is poor, sooner or later they will reach the melting point, and at that point they accept too
much MW energy in too short a time.
You can try this experiment with some other low melting material that does not decompose as easily, like for example KOH or NaOH. Put a pellet in a
sacrificial test tube and put this in a ceramic crucible. Heat it in the MW owen and report back your findings. 
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
At least i was not fully suprised by the explosion. I took distance and nessecary precautions.
I have a sort of sacrificial MW oven.
And I just didn't want to encourage anyone, or at least warn people who arn't aware of the danger from my previous suggestion.
Maybe ill do another test with far less power or some different conditions.
What a fine day for chemistry this is.
|
|
|
thereelstory
Harmless
Posts: 41
Registered: 18-1-2010
Member Is Offline
Mood: No Mood
|
|
thankyou for sharing your experience!
simply incredible and imaginative.
|
|
|
Hawkguy
Hazard to Others
Posts: 326
Registered: 10-10-2014
Location: British Columbia (Canada eh!)
Member Is Offline
Mood: Body is Ready
|
|
Could Ammonium Persulfate be thermally decomposed to yield Sulfur Trioxide? I'm exploring the route with Persulfates as I have no Sodium Bisulfate.
|
|
|
Oscilllator
National Hazard
Posts: 659
Registered: 8-10-2012
Location: The aqueous layer
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Hawkguy | Could Ammonium Persulfate be thermally decomposed to yield Sulfur Trioxide? I'm exploring the route with Persulfates as I have no Sodium Bisulfate.
|
Ammonium chloride reversibly decomposes at high temperature (338° according to wikipedia) to form NH3 and HCl. I would not be at all surprised if
this kind of thing happened with ammonium persulfate. You could try heating ammonium persulfate in a test tube and seeing if any ammonia is given off
to test this.
Sodium persulfate is available for me at the pool section of the local hardware store(Australia) so you could try there if you want a ammonium-free
alternative.
|
|
|
| Pages:
1
2 |
|












