| Pages:
1
..
69
70
71
72
73
..
104 |
JnPS
Hazard to Self

Posts: 90
Registered: 29-7-2016
Location: PA, USA
Member Is Offline
Mood: Umpolung
|
|
Can somebody download/share this article?
α,β-Unsaturated Aminoketones. VIII.1 Reaction of Primary Amines with 1,3-Diketones and Bromine Derivatives of Benzalacetophenone. Ethylene Imines
Norman H. Cromwell, Robert D. Babson, and Charles E. Harris
Journal of the American Chemical Society 1943 65 (3), 312-315
DOI: 10.1021/ja01243a003
It supposedly contains the full procedure for the paraphrased version (found in the Vogel's Organic chem book from the SM library) on a schiff base
prep formed from benzaldehyde and an methylamine. I'm still in the process of figuring out how to get access to acs publications trough my university
but would like to read this article during spring break to prepare for the procedure
|
|
|
gluon47
Hazard to Self
Posts: 81
Registered: 20-9-2015
Location: oceania
Member Is Offline
Mood: fluorinated and dying
|
|
Separation of potassium nitrite from nitrate
I've been following this procedure to produce potasium nitrite (link). I've just filtered off the lead(ii) oxide by product and discovered a fair amount of what looks like unreacted lead metal powder. This
means I'll have a fair amount of nitrate in my potassium nitrite solution.
I could evaporate down the solution to get crude potassium nitrite, but I want my product as pure as possible. I know methanol works quite well for
separating nitrite from nitrate, but unfortunately I don't have any.
How else could I go about separating and purifying my nitrite from nitrate?
any help would be greatly appreciated . .
reality is an illusion 
|
|
|
ficolas
Hazard to Others
Posts: 146
Registered: 14-5-2016
Member Is Offline
Mood: No Mood
|
|
Is there any viable oxidizer to form H2SO4 fron SO2 other than H2O2?
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
I've read about people doing it with nitric acid. The impurities will be much more volatile than the sulfuric acid and can be removed by distillation.
Can alcohols and carboxylic acids be brominated with antimony tribromide?
|
|
|
RogueRose
International Hazard
Posts: 1592
Registered: 16-6-2014
Member Is Offline
|
|
Depending on how much you need to convert you could look into an ozone (O3) generator. I've seen inexpensive chineese ones that sell for $30-50 that
supposedly produce 5g per hour = 120g / day and also ones that do 12g per hour = 288g per day. Some homemade units on youtube can produce A LOT
like 200g+ per hour but they run in the kilowatt rang for power consumption and are very dangerous (high voltage & high current) if not properly
used.
|
|
|
ficolas
Hazard to Others
Posts: 146
Registered: 14-5-2016
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by RogueRose  |
Depending on how much you need to convert you could look into an ozone (O3) generator. I've seen inexpensive chineese ones that sell for $30-50 that
supposedly produce 5g per hour = 120g / day and also ones that do 12g per hour = 288g per day. Some homemade units on youtube can produce A LOT
like 200g+ per hour but they run in the kilowatt rang for power consumption and are very dangerous (high voltage & high current) if not properly
used. |
Is the concentration of the ozone enough for it to be dissolved in cool water, so that I dont have to mix the two gases, I instead can just dissolve
them?
I think ill perform some experiments with thr SO2, scrubbing it (with NaOH?) to see if I can work with it safely.
Also, can I use some metal to burn the sulfur inside, or do I need to use glass because of corrosion?
[Edited on 13-3-2017 by ficolas]
|
|
|
Meltonium
Hazard to Self
Posts: 97
Registered: 23-9-2016
Location: Home in pajamas
Member Is Offline
Mood: Fluorinated
|
|
Free Radical Reaction
In a free radical reaction, is it better to use a shorter wavelength? For example, in free radical reaction using chlorine gas, would it be better to
use 200nm wavelength light or would it be better to use 400nm light?
[Edited on 3-17-2017 by Meltonium]
"If at first you don't succeed, fail, fail again."
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
You should use the lenght of wave suitable for your exact reaction/compound. AFAIK, the best wavelength for chlorine is 340 nm, maximum wavelength is
somewhere at 490 nm, which is where chlorine starts to absorb light, so 400 nm is perfectly fine.
|
|
|
fastbre4k
Harmless
Posts: 24
Registered: 29-11-2016
Member Is Offline
Mood: BENZIN
|
|
Phenylacetaldehyde
Will Phenylacetaldehyde Polymerize when stored solved?
|
|
|
Texium
|
Threads Merged
18-3-2017 at 11:41 |
j_sum1
Administrator
Posts: 6320
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
Capillary tubes for melting point determination
I am the proud owner of a Thiele tube. I also purchased a bunch of capillary tubes to go with it -- but without checking the diameter first. They
are very slender at 0.9mm ID (They actually look smaller than that.)
I think I need a wider tube just to get most crystalline substances in there.
What is the standard used?
And does anyone know of a good supplier? I spotted a pack of 100 2.4mm ID tubes that were going to set me back more than 50AUD by the time shipping
was included. That seems a bit excessive to me.
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
| Quote: Originally posted by j_sum1 | I am the proud owner of a Thiele tube. I also purchased a bunch of capillary tubes to go with it -- but without checking the diameter first. They
are very slender at 0.9mm ID (They actually look smaller than that.)
I think I need a wider tube just to get most crystalline substances in there.
What is the standard used?
And does anyone know of a good supplier? I spotted a pack of 100 2.4mm ID tubes that were going to set me back more than 50AUD by the time shipping
was included. That seems a bit excessive to me. |
That's a bit on the small side, but it doesn't actually sound too bad. I've got a tube of authentic melting point tubes sitting here and the OD is
~1.5mm. 2.4mm sounds a bit big, especially for the ID, and $50 is ridiculous(even considering it's aussie dollars).
I'd try running some melting points on a few well characterized compounds(benzoic acid, vanillin, etc.) and see how accurate your results are. If
they look good, those tubes will be fine, filling them is always a pain in the ass, I doubt it could get much harder . .
|
|
|
A Halogenated Substance
Hazard to Self
Posts: 68
Registered: 7-2-2017
Location: United States
Member Is Offline
Mood: Oxidizing due to extended exposure to oxygen
|
|
When boiling down a ferrous chloride solution, I added some steel wool to prevent the conversion of iron(II) to iron(III) by the air. The solution was
made from previously dissolved iron nails in HCl. However, as the boiling progressed, the solution went dark brown signifying the reaction to
iron(III) chloride.
The conversion did seem to start from the surface of the solution. Did heat help the conversion of iron(II) to iron (III) progress faster?
|
|
|
Geocachmaster
Hazard to Others
Posts: 146
Registered: 5-3-2016
Location: Maine, USA
Member Is Offline
Mood: Corroded, just like my spatulas
|
|
CaCl2 and ethyl acetate?
About an hour ago I added a few scoops of CaCl2 to ~50ml of ethyl acetate thinking that I would leave it to dry overnight. I was so sure that CaCl2
was compatable because I had read somewhere that ethanol could be removed from EtAc with calcium chloride. Upon reading this I now am doubtful because it does not say calcium chloride can be used. I wanted some dry EtAc for tomorrow so...
Will CaCl2 work for drying ethyl acetate or should I use another drying agent?
|
|
|
mayko
International Hazard
Posts: 1218
Registered: 17-1-2013
Location: Carrboro, NC
Member Is Offline
Mood: anomalous (Euclid class)
|
|
I've made a couple pounds of Fe2O3 from precipitating an iron solution with base (ammonia/sodium carbonate), oxidizing with peroxide, and heating to
dryness. The thing is, the red powder seems to be strongly magnetic! I'm still scratching my head about this; the solutions had been filtered to
remove suspended iron particles. The best I've come up with is some leaves might have fallen into the solid as it was being heated down over charcoal
outside, and the carbon reduced the oxide to metallic iron. However, the magnetism is spread throughout the powder, and I haven't been able to
mechanically separate it into magnetic and non-magnetic portions. Any idea what's going on?
al-khemie is not a terrorist organization
"Chemicals, chemicals... I need chemicals!" - George Hayduke
"Wubbalubba dub-dub!" - Rick Sanchez
|
|
|
Liamatpm
Harmless
Posts: 48
Registered: 7-12-2016
Location: Maine
Member Is Offline
Mood: Using the process of cellular respiration to stay (barely) alive
|
|
Yes it should work. And if it doesn't it was worth a try.
|
|
|
A Halogenated Substance
Hazard to Self
Posts: 68
Registered: 7-2-2017
Location: United States
Member Is Offline
Mood: Oxidizing due to extended exposure to oxygen
|
|
| Quote: Originally posted by mayko | | I've made a couple pounds of Fe2O3 from precipitating an iron solution with base (ammonia/sodium carbonate), oxidizing with peroxide, and heating to
dryness. The thing is, the red powder seems to be strongly magnetic! I'm still scratching my head about this; the solutions had been filtered to
remove suspended iron particles. The best I've come up with is some leaves might have fallen into the solid as it was being heated down over charcoal
outside, and the carbon reduced the oxide to metallic iron. However, the magnetism is spread throughout the powder, and I haven't been able to
mechanically separate it into magnetic and non-magnetic portions. Any idea what's going on? |
Do you think that you could've produced magnetite?
http://www.sciencemadness.org/smwiki/index.php/Iron(II,III)_oxide
|
|
|
A Halogenated Substance
Hazard to Self
Posts: 68
Registered: 7-2-2017
Location: United States
Member Is Offline
Mood: Oxidizing due to extended exposure to oxygen
|
|
| Quote: Originally posted by Geocachmaster | About an hour ago I added a few scoops of CaCl2 to ~50ml of ethyl acetate thinking that I would leave it to dry overnight. I was so sure that CaCl2
was compatable because I had read somewhere that ethanol could be removed from EtAc with calcium chloride. Upon reading this I now am doubtful because it does not say calcium chloride can be used. I wanted some dry EtAc for tomorrow so...
Will CaCl2 work for drying ethyl acetate or should I use another drying agent?
|
According to the Sciencemadness wiki, calcium chloride is compatible with esters so it should work.
http://www.sciencemadness.org/smwiki/index.php/Drying_solven...
|
|
|
Sulaiman
International Hazard
Posts: 3692
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
Opinions on ptfe vs boiling sulphuric acid
I intend to distill some drain-unblocker grade sulphuric acid (93 - 96 %w/w) to get some clean, nearer to azeotropic, sulphuric acid.
I intend to use a 3-neck 500 ml flask with a thermometer pocket in one of the side necks.
Unfortunately the adapter needs a few wraps of ptfe plumbers tape to fit properly.
Azeotropic sulphuric acid boils at 337 oC ... https://en.wikipedia.org/wiki/Azeotrope_tables
ptfe melts at 327 oC ... https://en.wikipedia.org/wiki/Polytetrafluoroethylene
What are my chances of success ?
If the ptfe does melt, what are the chances of separating the thermometer pocket and flask ?
 
I modified from the first picture to the second to allow air cooling of the joint, as suggested below ... thanks yobbo II
[Edited on 23-3-2017 by Sulaiman]
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
yobbo II
National Hazard
Posts: 762
Registered: 28-3-2016
Member Is Offline
Mood: No Mood
|
|
Set up something that blows air at the teflon area
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
You can't get azeotrope by distillation, unless you have a fractioning column.
And second: why would you need to measure the temperature of the boiling acid and not the temperature of vapor?
|
|
|
Sulaiman
International Hazard
Posts: 3692
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Offline
|
|
This is pre-planning for when I finish my diy heating mantle, some weeks ahead
I intend to use a fractionating column to get nearer to azeotropic, not azeotropic
... I expect that to be too difficult.
I want to monitor the temperature at the top and bottom of the column so that I can estimate my still performance at higher temperatures.
Later I want to use the same (or very similar) still setup to distill my mercury - at even higher temperatures.
... because I can 
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
Geocachmaster
Hazard to Others
Posts: 146
Registered: 5-3-2016
Location: Maine, USA
Member Is Offline
Mood: Corroded, just like my spatulas
|
|
Acetyl chloride?
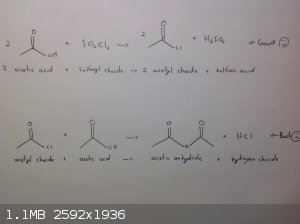
I've read on the SM wiki and on the acetic anhydride thread that sulfuryl chloride will react with acetate salts to form acetyl chloride. Reaction
with acetic acid instead has the advantage of being a totally liquid reaction mixture. Meaning stirring, cooling and temperature readings would all be
much easier. However I cannot find any references to the reaction of acetic acid and sulfuryl chloride to produce acetyl chloride. I don't think that
the bottom reaction would matter because the reverse reaction is used to make acetyl chloride. Of all the components acetyl chloride has the lowest
boiling point by 20C so fractional distillation should drive the reaction to completion. I think it makes sense if acetates can be used.
Does anyone have information or knowledge about a reaction between sulfuryl chloride and acetic acid to produce acetyl chloride?
Thanks for your time
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
I really doubt you can get acetyl chloride from a sole acetic acid. That's not a thionyl chloride, I'd say reaction products would be (AcO)SO2 + HCl,
in case you manage to get something reacted..
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
IIRC sulfuryl chloride does not react readily as an electrophile. Acetate is a weak electrophile (weaker than Cl- in water) and probably won't react
with it. SO2Cl2/HOBt generates a reactive sulfonyl halide, and tosyl chloride has been used to make Ac2O from acetic acid, so that might work, but
you'd need benzotriazole.
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
What kind of needles do you use for handling bromine and/or 37% hydrochloric acid with a glass syringe?
|
|
|
| Pages:
1
..
69
70
71
72
73
..
104 |










