| Pages:
1
..
4
5
6
7
8 |
samarium14
National Hazard

Posts: 661
Registered: 22-3-2019
Location: Georgia
Member Is Offline
|
|
Hi. These photos are better. Last one is new (cadmium violurate). Unfortunately the indium violurate powder was spilled. I have so little right now
that it is nonsense to stuff it in a vial.
       
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Amazing work vano!
Haven't seen lead mentioned, so here is some. Made from about 0.11 g of the sodium salt (dihydrate) and about 0.17 g of lead acetate trihydrate, from
mixing the two as solutions (added lead to the violurate). Instant precipitate from the warm solution, but no complete discoloration. Filtered,
washing water was colorless. A portion was dried at about 180 °C on a hotplate.
Left is the tetra/pentahydrate (still a bit wet), right anhydrous
 
Didn't find much information, and some conflicting on the hydration. Can't access the original source so I'll attach the scifinder screenshots.
 
we apologize for the inconvenience
|
|
|
samarium14
National Hazard
Posts: 661
Registered: 22-3-2019
Location: Georgia
Member Is Offline
|
|
Thanks Diachrynic. Nice work.
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Yttrium is generally not very interesting, but it's violurate is brightly yellow.
I dissolved about 0.5-0.7 g of violuric acid monohydrate (mine was freshly made and still wet so I don't know exactly, it's an excess of over 3:1 to
the yttrium used however) in 50 mL of boiling 95% ethanol - had some insoluble grey-brown stuff so I filtered that off. To that solution was added
about 0.25 g of YCl3 (it should be noted that YCl3 is very hygroscopic and mine was slightly wet so this amount is also lower
than I measured out here) in about 1 mL of water to the boiling ethanol solution while stirring. It turned yellow and after about a minute of strong
stirring and boiling the solution became turbid, it was boiled for five minutes longer and left to cool until luke warm. The yellow precipitate was
vacuum filtered off and washed with some 95% ethanol. The solution that passes through is slightly yellow and deposits only small amounts of
additional precipitate with more yttrium chloride solution. The solid was then air dried. Yield was about 0.166 g. (Because this was all done a bit
sloppy I didn't bother to calculate a % yield.)
![[YVio2(H2O)2]Vio+6H2O.jpg - 2MB](https://www.sciencemadness.org/whisper/files.php?pid=657439&aid=87825)
The product is of the composition [Y(Vio)2(H2O)2]Vio · 6 H2O where HVio is violuric acid.
The preparation comes from Gad, A. A. M., Farag, I. S. A., & Awadallah, R. M. (1992). Synthesis and Characterisation of Scandium (III)-, Yttrium
(III)-, and Lanthanum (III) Violurate Complexes. Crystal Research and Technology, 27(2), 201–210. doi:10.1002/crat.2170270210
Attachment: gad1992.pdf (421kB)
This file has been downloaded 416 times
[Edited on 2-4-2021 by Diachrynic]
we apologize for the inconvenience
|
|
|
samarium14
National Hazard
Posts: 661
Registered: 22-3-2019
Location: Georgia
Member Is Offline
|
|
Nice. Now there are two yellow violurates Sc and Y. Where did you buy yttrium chloride?
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
I happened to get about 6 kg (it was one big solid brick) of it when it was supposed be disposed of, so I saved it from being just thrown out, I don't
actually know where to buy it.
[Edited on 2-4-2021 by Diachrynic]
we apologize for the inconvenience
|
|
|
samarium14
National Hazard
Posts: 661
Registered: 22-3-2019
Location: Georgia
Member Is Offline
|
|
6 kg is too much, good source. I too have bought solid brick chemicals very cheaply.
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
I found no literature on bismuth violurate, so here is a small test tube scale experiment. It seems fairly soluble.
Some bismuth nitrate pentahydrate was dissolved in some water with some sulfuric acid (bismuth hydrolyses to basic salts with annoying ease...), to
this some sodium violurate solution was added. The color turned orange. When acetic acid and sodium acetate were added, the color became purple.
Whether this is because the sodium is competing with the bismuth or something else, I am not sure, the color is somewhat different from pure sodium
violurate however, which is more purple. If anyone wants to prepare the solid, I would suggest using a bismuth oxide/hydroxide and the free acid since
I didn't get precipitation.
 
we apologize for the inconvenience
|
|
|
h0lx
Hazard to Self
Posts: 69
Registered: 31-12-2005
Member Is Offline
|
|
ortho-toluidine violurate, this one I did as a proof of concept in a very small unmeasured scale, I will now make more to vial up.

|
|
|
RustyShackleford
Hazard to Others
Posts: 202
Registered: 10-12-2020
Location: Northern Europe
Member Is Offline
|
|
Update on the subject of violuric acid:
Thanks to the help of PoorMans Chemist (who i sent some violuric and barbituric acid to) videos about the subject have been produced, aswell as many
new salts for our collective viewing pleasure.
Especially big thanks to him for producing the Thallium and Uranyl salts, super cool to see!
Also thanks to user H0lx for giving me the idea to put these tables together, aswell as making many of the organic salts of violuric acid (pyridine,
ethanolamine, methylamine, ethylenediamine, o-MePhNH3, Guanidine and Aminoguanidine)
I decided to put together 2 new tables to better show all (i think) organic salts created thus far, aswell as update the periodic table. Unfortunately
i had to cut up the organic one due to file size limit
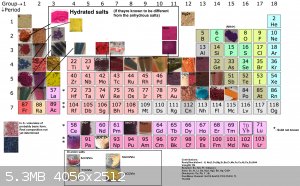
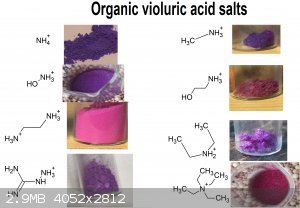
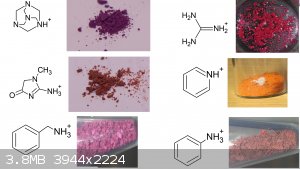
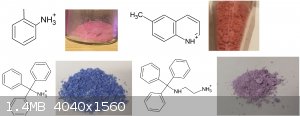

If you can find a pattern in these colors, please inform me.
If youd like to watch the videos PoorMans Chemist created:
Preparation of Ba, Cs, Tl violurates:
https://www.bitchute.com/video/427j5UDiP7oQ/
Preparation of violuric from barbituric:
https://www.bitchute.com/video/pPvACwLjOdUw/
Preparation of a few organoammonium violurates:
https://www.bitchute.com/video/VCevp82sbNIC/
His youtube channel, community page contains several pictures of the salts aswell as of the process of producing them:
https://www.youtube.com/c/PoorMansChemist/
[Edited on 6-7-2021 by RustyShackleford]
|
|
|
j_sum1
Administrator
Posts: 6374
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
I am loving this project -- at least watching from the side lines.
About your periodic table graphic, surely it is Sc3+?
|
|
|
samarium14
National Hazard
Posts: 661
Registered: 22-3-2019
Location: Georgia
Member Is Offline
|
|
I love this project too. Yes it's really scandium salt. I don't have much scandium because it has high price. I bought 1 gram scandium and i used very
small amount for this reaction.

|
|
|
RustyShackleford
Hazard to Others
Posts: 202
Registered: 10-12-2020
Location: Northern Europe
Member Is Offline
|
|
Quote: Originally posted by j_sum1  | I am loving this project -- at least watching from the side lines.
About your periodic table graphic, surely it is Sc3+? |
damn, and i thought i fixed the errors in the image. Ofc it is and should have been obvious to me. ill fix it in the next update when ive done the
Al3+ salt and Ti3+ salt. I already tried Al twice but with no success.
Im having trouble getting a pure sample of Al(OH)3 to react w the acid. precipitating the hydroxide and washing it didnt work, and using hydrolysed
then dried AlCl3 gave some wierd product that evaporated to a white solid (purple/brown in solution), very strange.
[Edited on 7-7-2021 by RustyShackleford]
|
|
|
samarium14
National Hazard
Posts: 661
Registered: 22-3-2019
Location: Georgia
Member Is Offline
|
|
It would be interesting to make oxime from thiobarbituric acid a violuric acid analogue. I wonder how different the colors will be from violurates.
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
| Quote: Originally posted by RustyShackleford |
...
damn, and i thought i fixed the errors in the image. Ofc it is and should have been obvious to me. ill fix it in the next update when ive done the
Al3+ salt and Ti3+ salt. I already tried Al twice but with no success.
Im having trouble getting a pure sample of Al(OH)3 to react w the acid. precipitating the hydroxide and washing it didnt work, and using hydrolysed
then dried AlCl3 gave some wierd product that evaporated to a white solid (purple/brown in solution), very strange.
[Edited on 7-7-2021 by RustyShackleford] |
I tried the Al3+ salt too. Solution and crystals are fuchsia-like, similar to a number of other violurates:
 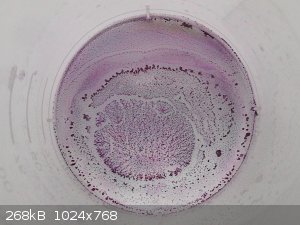
Preparation via Al2(CO3)3 from alum with sodium bicarbonate solution and filtrating/rinsing twice. Stir the carbonate with the violurate for 1 hour on
1200rpm at 70C. Let stand for a day and using a pipette to suck off the solution.
I don't exclude that a larger portion of the violuric acid hasn't even reacted.
|
|
|
DraconicAcid
International Hazard
Posts: 4413
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Alum and sodium bicarbonate don't give you aluminum carbonate- it gives aluminum hydroxide.
Still, a pretty colour.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
| Quote: Originally posted by Diachrynic | Yttrium is generally not very interesting, but it's violurate is brightly yellow.
I dissolved about 0.5-0.7 g of violuric acid monohydrate (mine was freshly made and still wet so I don't know exactly, it's an excess of over 3:1 to
the yttrium used however) in 50 mL of boiling 95% ethanol - had some insoluble grey-brown stuff so I filtered that off. To that solution was added
about 0.25 g of YCl3 (it should be noted that YCl3 is very hygroscopic and mine was slightly wet so this amount is also lower
than I measured out here) in about 1 mL of water to the boiling ethanol solution while stirring. It turned yellow and after about a minute of strong
stirring and boiling the solution became turbid, it was boiled for five minutes longer and left to cool until luke warm. The yellow precipitate was
vacuum filtered off and washed with some 95% ethanol. The solution that passes through is slightly yellow and deposits only small amounts of
additional precipitate with more yttrium chloride solution. The solid was then air dried. Yield was about 0.166 g. (Because this was all done a bit
sloppy I didn't bother to calculate a % yield.)
The product is of the composition [Y(Vio)2(H2O)2]Vio · 6 H2O where HVio is violuric acid.
The preparation comes from Gad, A. A. M., Farag, I. S. A., & Awadallah, R. M. (1992). Synthesis and Characterisation of Scandium (III)-, Yttrium
(III)-, and Lanthanum (III) Violurate Complexes. Crystal Research and Technology, 27(2), 201–210. doi:10.1002/crat.2170270210
[Edited on 2-4-2021 by Diachrynic] |
I have some doubt whether you really got the salt you say you got, [Y(Vio)2(H2O)2]Vio · 6 H2O. The rare
earths tend to form yellow violurate salts which contain chloride when prepared from rare earth chlorides, so it may have played a trick on you.
I prepared a yellow salt from EuCl3.6H2O (pictured). However, with Sm2(CO3)3 and Pr(OH)3 I obtained dark red salts.

|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
Okay, I wasn't aware of that.
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
Cerium violurate
0.374g of (NH4)4Ce(SO4)4 were dissolved in water and Ce(OH)4 was precipitated by adding an excess of NaOH solution. The precipitate was washed 4 times
and then scraped of the filter and added to a warm (pink/lilac) solution of 0.298g of violuric acid. The light yellow precipitate of Ce(OH)4 turned
dark orange immediately but the solution retained its colour of dissolved violuric acid.
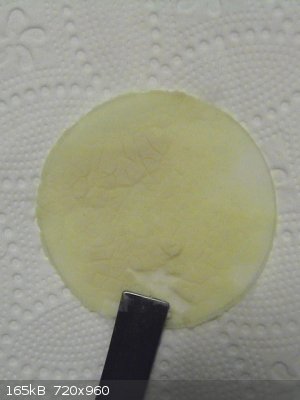 
This is a bit mysterious, since the Ce(OH)4 was present in excess (0.300g (NH4)4Ce(SO4)4 would be stoichiometric, assuming no losses). It did react
with the violuric acid, but it did not dissolve nor use up all the dissolved violuric acid. I presume that a basic Ce-salt has formed, like
Ce(OH)2(vio)2.
|
|
|
woelen
Super Administrator
Posts: 8082
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
I finally ordered some barbituric acid (from S3 chemicals), so that I also can jump in in this thread. I certainly want to try things, I want to try
making mix-salts with different metal ion. Sometimes such mix-salts can have really surprising colors, due to electronic interactions between ions in
a (closely packed) lattice. But first I'll try some of the variations in this thread, just to check my violuric acid, which I first need to prepare.
|
|
|
RustyShackleford
Hazard to Others
Posts: 202
Registered: 10-12-2020
Location: Northern Europe
Member Is Offline
|
|
| Quote: Originally posted by woelen | | I finally ordered some barbituric acid (from S3 chemicals), so that I also can jump in in this thread. I certainly want to try things, I want to try
making mix-salts with different metal ion. Sometimes such mix-salts can have really surprising colors, due to electronic interactions between ions in
a (closely packed) lattice. But first I'll try some of the variations in this thread, just to check my violuric acid, which I first need to prepare.
|
Glad to hear! if you need any help send me a DM. I have a good amount of experience doing that prep and making salts with it , 24 so far 
|
|
|
woelen
Super Administrator
Posts: 8082
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
My barbituric acid arrived
Next weekend I want to make some violuric acid.
@RustyShackleford: You posted this link https://illumina-chemie.de/viewtopic.php?t=5502
I want to go through sodium violurate instead of making violuric acid directly (the latter only has ~40% yield, while the other two-step proces
through Na-violurate has a combined ~70% yield). How was your experience with this preparation? Any special remarks or pitfalls. I have 50 grams of
barbituric acid, so I should have enough for a few shots, but of course I want to have it right at first shot 
|
|
|
RustyShackleford
Hazard to Others
Posts: 202
Registered: 10-12-2020
Location: Northern Europe
Member Is Offline
|
|
| Quote: Originally posted by woelen | My barbituric acid arrived
Next weekend I want to make some violuric acid.
@RustyShackleford: You posted this link https://illumina-chemie.de/viewtopic.php?t=5502
I want to go through sodium violurate instead of making violuric acid directly (the latter only has ~40% yield, while the other two-step proces
through Na-violurate has a combined ~70% yield). How was your experience with this preparation? Any special remarks or pitfalls. I have 50 grams of
barbituric acid, so I should have enough for a few shots, but of course I want to have it right at first shot |
[Barbituric acid->Na Violurate -> Violuric acid] is the route ive done 4 times now, every time its gone smoothly. The yield has been VERY good,
starting with 86% overall and climbing to 92% most recently.
The 2 step prep as written on illumina has worked very well, the only change i made is to disregard being accurate with the pH. I simply just added
the 1g of NaOH and 3 ml acetic acid, and later 2.5g NaOH, which doesnt actually end up at the pH the prep says,but it seemingly doesnt even matter.
Also, for the Na violurate -> violuric acid step, i stir it 1h with 15% HCl, then another hour with fresh 15% HCl. This is to leech out more Na
ions which otherwise could contaminate the violuric acid. After that, i filter it off and dry in an NaOH dessicator (puts out a lot of HCl fumes while
drying, can be washed w cold isopropanol to lessen the fumes at a cost of 4-5% in yield)
When its dessicated, its good to dry it further in oven/on hotplate @140C so you can be really accurate with stoiciometry when using it.
If you want to recrystallize the violuric acid (i never did), i think a suitable solvent would be 5% HCl
[Edited on 29-7-2021 by RustyShackleford]
|
|
|
woelen
Super Administrator
Posts: 8082
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
I roughly used the route, described on Illumina, but I did not use any NaCl. That adds another ion (chloride) in the mix and I want to have as little
as possible of other anions in the mix.
I proceeded as follows:
- Dissolve 3.25 grams of barbituric acid in 50 ml of distilled water. Heat the water to near boiling, so that all of the barbituric acid dissolves.
- Dissolve 1.92 grams of NaNO2 in 6 ml of water.
- Add the solution of NaNO2 to the solution of barbituric acid, while the latter is on a stirrer with a stir bar in it. Rinse all NaNO2 from the
beaker with an addition few ml of water --> As soon as the NaNO2 goes into the solution of barbituric acid, the liquid becomes dark purple. In the
next few minutes, solid sodium violurate starts to settle, despite the stirring.
- In a separate beaker, put 4 ml of CH3COOH, 4 ml of water and add 1 gram of NaOH. A violent reaction occurs, all NaOH dissolves. Pour this hot liquid
into the liquid with the barbituric acid and NaNO2, while still stirring. Keep on stirring for 20 minutes or so, when no changes occur anymore.
- Dissolve 2.5 grams of NaOH in 5 ml of water and add this solution to the beaker with the sodium violurate. Keep stirring and at the same time, heat
the beaker till near boiling --> The solid sodium violurate does not dissolve. Even at boiling, it remains suspended in the liquid. When stirring
stops, then the solid settles in a few tens of seconds into a layer of 1 cm thickness.
- Finally, allow the liquid in the beaker to cool down and then put it in a fridge at 5 C or so.
Now, the beaker still is in the fridge, I'll leave it there for an hour or so and then I'll see how much sodium violurate I can separate. It looks
quite good though, I see a lot of compact precipitate.
I also measured the pH of the liquid at the end, after adding the 2.5 grams of NaOH. The liquid has a pH of 11 to 12, according to my pH-paper. This
is quite alkaline. It does not feel slippery to the skin though.
------------------------------------------------------------
If I indeed can separate a nice amount of sodium violurate, then I'll try exactly the same experiment, with the same molar amounts with the potassium
ion instead of sodium ion. I have KNO2, KOH and then can use the same amounts of barbituric acid, the same molar amount of KNO2 and the same molar
amounts of KOH. That should give me K-violurate, without any chloride in it.
I also have Ba(NO2)2 and Ba(OH)2, so maybe I even try the experiment with Ba in half the molar amount in barium, but I expect that experiment to be
more cumbersome. Ba(NO2)2 and Ba(OH)2 are not that well soluble, and solutions tend to get cloudy if they are somewhat alkaline (due to absorption of
CO2 from air and subsequent precipitation of BaCO3).
More will follow, and of course, I make pictures and videos of all steps I take
|
|
|
woelen
Super Administrator
Posts: 8082
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
I can report that the production of sodium violurate succeeded very well with the above procedure. I took the beaker from the fridge and filtered the
solution. This leads to a paste. This paste I rinsed one time with a small amount of distilled water and then I again filtered until I obtained a
paste again. This I dried in a watch glass on a warm place (50 C or so), while occasionally stirring the material and turning it around. I allowed it
to dry for 36 hours, but after 24 hours it looked dry already.
I have done my best to have as little as possible of mechanical losses. I had a yield of 4.53 grams of solid material, a free flowing powder with a
deep red, somewhat pink/purple color. It really has a beautiful very bright color, which I could capture in a picture quite well. Pictures will follow
soon, together with a webpage.
My percentage yield is a little lower than reported in the first post (there, a yield of 9.85 grams is reported from double amounts, so, based on that
I should have 4.93 grams or so). But the difference is not dramatic, so I am quite happy with my result. My result is definitely free of chloride and
that may be helpful when experimenting with coordination complexes.
I also did a few experiments with acids and bases:
- Adding the solid to 10% H2SO4 leads to the solid turning orange/red instead of the bright purple/red and it dissolves, giving a colorless solution.
- Adding the solid to 10% NaOH leads to dissolving of the solid, just like in water, but the solution is not red with a purple tinge, but it is more
orange/red. Still a bright color, but definitely different from what you get, when the solid is dissolved in water. I already noticed that at high pH,
the solution tends to shift to orange.
- I added excess solid NaOH to the colorless solution in dilute H2SO4. Around the granules of NaOH you see pale orange liquid. The solid NaOH
dissolves, producing a lot of heat. At the lower, strongly alkaline layer, the liquid becomes orange, then there is a very dark purple/blue layer and
on top of that is a pink acidic layer. On shaking all of the liquid, the final color becomes red with an orange hue, just like the solution, obtained
when dissolving sodium violurate in dilute NaOH.
[Edited on 3-8-21 by woelen]
|
|
|
| Pages:
1
..
4
5
6
7
8 |









