| Pages:
1
..
4
5
6
7 |
Motherload
Hazard to Others

Posts: 245
Registered: 12-8-2012
Location: Sewer
Member Is Offline
Mood: Shitty
|
|
If you start adding PE at a temp that's too low .... The PE builds up and not nitrated to PETN. So when the temp rises to the nitration temp .... All
the surplus PE wants to get nitrated at the same time causing hot spots and spikes which is probably what results in runaway signs.
"Chance favours the prepared mind"
"Fuck It !! We'll do it live !!"
|
|
|
ONLIASHISH
Harmless
Posts: 3
Registered: 4-3-2013
Member Is Offline
Mood: Learner
|
|
PETN Synth
Hi Motherload, Thank you for your help,I guessed the same reason as you have mentioned. I did lot of search found I should have done nitration at 15C
to 20C very slowly. I have recrystallize impure PETN with acetone at 50C and then filtered after adding water yield is 12g(instead of 22.1g). Learnt
something from mistake 
|
|
|
ONLIASHISH
Harmless
Posts: 3
Registered: 4-3-2013
Member Is Offline
Mood: Learner
|
|
PETN Synth
Hi 10digit,Thank you for your help. I had pure acids without any other impurities other than water. I had done other synthesis(Nitroglycerine,nitro
urea, GC etc) with same acids . The reason given by Mother load is more accurate for my case.
|
|
|
Hellbat
Harmless
Posts: 16
Registered: 23-2-2014
Member Is Offline
Mood: No Mood
|
|
Question about PETN synthesis
Hi, I have found a synthesis description for PETN that uses 65% nitric acid instead of fuming nitric acid: http://www.powerlabs.org/chemlabs/petn.htm
But on many other pages I've read that nitrating PE with the mixed acid method mostly yields Pentaerythritoltrinitrate (PETRIN). It
seems to be soluble in acetone just as well, so the purification by recrystalization would not remove it. Maybe, this guy actually made the trinitrate
without knowing? It's a strong explosive as well.
I've actually found a german synthesis description for PETRIN which was nearly the same procedure as on the website I linked.
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
| Quote: | | Maybe, this guy actually made the trinitrate without knowing? |
I have the feeling that when Sam Barros 'makes something' he knows exactly what it is he's made!
|
|
|
Hellbat
Harmless
Posts: 16
Registered: 23-2-2014
Member Is Offline
Mood: No Mood
|
|
But does anyone have accurate information about that? According to german wikipedia, if PE is nitrated with mixed acid, mostly the trinitrate is
formed...
So, either wikipedia or Sam Barros must be wrong.
|
|
|
Bert
Super Administrator
Posts: 2821
Registered: 12-3-2004
Member Is Offline
Mood: " I think we are all going to die. I think that love is an illusion. We are flawed, my darling".
|
|
Why don't you do an experiment and verify if Wikipedia is wrong, and report back.
-------
The method and ratios work. I have tried the version of this originally posted by "Boomer" that Sam apparently either directly copied or used the same
information (from Thadeus Urbanski Vol. IV) to derive. Should give 90% + yields with care and the correct grade of pentaerythritol.
| Quote: |
Verified efficient method of preparing PETN from diluted HNO3
use
32,7 ml of 70% HNO3 (75% excess)
18,9 ml of 96% H2SO4
10,0 g of Pentaerythrite
------------------------
or
34,0 ml of 65% HNO3 (66% excess)
24,8 ml of 96% H2SO4
10,0 g of Pentaerythrite
------------------------
or
34,8 ml of 58% HNO3 (48% excess)
36,3 ml of 96% H2SO4
10,0 g of Pentaerythrite
------------------------
Nitration:
1. Cool nitration mixture during mixing of acids, thus minimalize even minimal decomposition of HNO3. Nitration mixture must be cooled before
nitration process to temperature of 10°C.
2. While stiring, add pentaerythritee in small portions (1-2g) to nitration mixture, always after previous batch is dissolved. Nitration mixture
gradually thicken as PETN forming in solution.
3. Constantly monitor reaction temperature and maintain it in 10-15°C range. Interval of adding pentaerytritol conform to reaction temperature, must
not rise over 15°C, leave beaker in cold water.
4. Stir with mixture for next 5 minutes after all pentaerythrite is added and dissolved. Mixture is now thick, but stirring is going well. During
nitration process must not be developed any brown fumes of NOx!
5. Now put beaker with mixture into water bath and maintain temperature at 50°C, continuously stir with mixture. During 20 minutes at this
temperature, all of possible sulfoesters come into PETN for maximal yield of nitration.
6. While maintaining mixture at higher temperature, mixture must be monitored for developing of NOx fumes. Only light brown colour can be in shrouded
beaker. Raised development of NOx pointing to higher temperatures used (even during previous nitration) or insufficient chmemicals purity and further
heating may end up in uncontrolled reaction and oxidation of formed PETN. In this case it is better to do not heat at all and end just after nitration
(if brown fumes apperas during nitration), the yield will be lower. In case of accidentally runaway reaction during heating, immedialtely pour mixture
into cold water, don't try stop reaction by cooling beaker, it will not help.
7. When heating after 20 minutes pass off, pour reaction mixture into cold water and follow standart procedure of filtration, neutralization and
purifying of PETN.
Yield was 22,1g of PETN from 10g of pentaerythrite, ~95% of theoretical yield (with 65% HNO3 used).
This procedure is result of my research of most effective method preparing PETN from diluted HNO3. Acid ratios are precisely calculated on data from
PETN nitration graph published by T. Urbanski in his book vol. IV. Generaly said, PETN is forming to maximum 30% of water portion in nitration
mixture. But, when also H2SO4 is contained in nitration mixture, minimum ammount of water must be keep, or oxidation with small yields occur. With
this acid ratios 20% of H2O is minimum. So, this nitration mixture has 20% of water on nitration start and 30% at the end, area for most effective
nitration, but again only with this ratios. For other acid ratios must be all recalculated. Excess of HNO3 is used to controll ammount of reaction
water. Only ammount of nitration mixture can be extended for lower mixture thickness, but it lower utilization of acids and overall efficiency. But it
isn't necessary.
I hope there is no major mistakes in my translation to english ;-)
|
I am going to merge your thread with an existing PETN thread-
http://www.sciencemadness.org/talk/viewthread.php?tid=10413&...
And you might want to look at the alternative method posted by Rosco Bodine:
http://www.sciencemadness.org/talk/viewthread.php?tid=6151#p...
[Edited on 11-1-2016 by Bert]
Rapopart’s Rules for critical commentary:
1. Attempt to re-express your target’s position so clearly, vividly and fairly that your target says: “Thanks, I wish I’d thought of putting it
that way.”
2. List any points of agreement (especially if they are not matters of general or widespread agreement).
3. Mention anything you have learned from your target.
4. Only then are you permitted to say so much as a word of rebuttal or criticism.
Anatol Rapoport was a Russian-born American mathematical psychologist (1911-2007).
|
|
|
Bert
|
Threads Merged
11-1-2016 at 07:23 |
NeonPulse
Hazard to Others
Posts: 417
Registered: 29-6-2013
Location: The other end of the internet.
Member Is Offline
Mood: Isolated from Reality! For Real this time....
|
|
I'll too vouch for roscoes method. It is the best synthesis I've tried. No problem in producing high yielding high quality PETN that is easily
neutralised and Recrystallized. No heating needed and no Dangerous fuming. You do need High strength clear fuming nitric though. But the trouble is
worth it. The end product does seem superior to mixed acid PETN
|
|
|
gnitseretni
Hazard to Others
Posts: 283
Registered: 5-1-2007
Location: Colombia
Member Is Offline
Mood: No Mood
|
|
I don't think the trouble is worth it. Only if you're specifically after PETN for some reason. Otherwise, just go the mixed acids route (assuming
that's the easier route for you). You won't notice the difference between a 7k or 7.5k velocity explosive.
I precool my acids and I add my PE while the acids are still below freezin, and I add it pretty fast. It does tend to heat up pretty fast after a
certain point but as long as you are prepared and have plenty of ice available you'll be fine.
|
|
|
Microtek
National Hazard
Posts: 920
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
The superiority of the product has more to do with stability than with performance, I think. It is very difficult to remove the last traces of
sulfuric acid from the product, and this makes it unstable, particularly at elevated temperatures.
|
|
|
Etanol
Hazard to Others
Posts: 220
Registered: 27-2-2012
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Bert  |
The method and ratios work. I have tried the version of this originally posted by "Boomer" that Sam apparently either directly copied or used the same
information (from Thadeus Urbanski Vol. IV) to derive. Should give 90% + yields with care and the correct grade of pentaerythritol.
|
It seems the diagram in the 4th volume is a lie. 90% PETN output is also formed in mixtures with a small water content in which, according to the
diagram, it should not form.
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1447
Registered: 2-9-2014
Location: Tel Aviv University
Member Is Offline
Mood: old jew
|
|
Since recently there have been ambiguities regarding the synthesis of PETN, I am attaching a repeatedly proven method of preparation in graphic form
to the relevant thread....
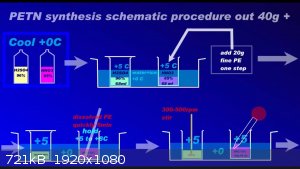 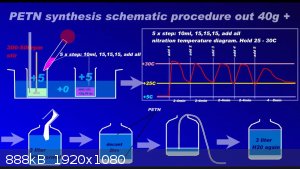 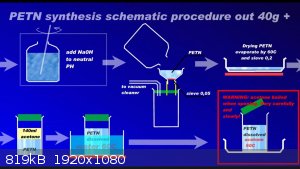
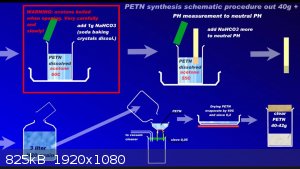
Development of primarily - secondary substances: CHP (2015) neutral CHP and Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024) Diper
60 (2025)
|
|
|
DennyDevHE77
Hazard to Others
Posts: 168
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
Has anyone tested the product above on a chromatograph? How does it match PETN obtained with pure 98% nitric acid?
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1447
Registered: 2-9-2014
Location: Tel Aviv University
Member Is Offline
Mood: old jew
|
|
Using 98% HNO3 can cause more trouble than good. But that's just my guess. Using 98% HNO3 has not been tested in this process. Chromatography was not
used for measurement PETN from graphic guide process. But the result is stable and quality PETN. Repeatedly. 20g PE can be added to 65% HNO3 at once.
Without dramatic increase of temperature. PE is completely soluble in HNO3 65% by picture.
Development of primarily - secondary substances: CHP (2015) neutral CHP and Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024) Diper
60 (2025)
|
|
|
DennyDevHE77
Hazard to Others
Posts: 168
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
That's not the point. When using a mixture of nitric and sulfuric acid, it also partially converts pentaerythritol into mixed sulfoesters. When heated
to 50-60C, nitric acid will convert these esters to PETN. But some esters (I think pentaerythritol dinitrate-disulfate) are still not completely
converted to PETN.
I used to (and still do sometimes) boil PETNs in 1% baking soda solution to remove them. But this resulted in 5-10% yield loss, later an ethanol
member advised me to "cook" PETN in 85-90C soda solution, without boiling, and it seemed to work.
That's the upside of nitrating 90-99% nitric acid. PETN is obtained with a slightly higher yield (up to 98%), and most importantly it is formed
immediately pure. All that is needed afterwards is to remove the acid in acetone, and that's all.
|
|
|
ManyInterests
National Hazard
Posts: 968
Registered: 19-5-2019
Member Is Offline
|
|
I am making some ammonium nitrate since I managed to find a place that sells ammonium carbonate for relatively cheap and am turning some calcium
nitrate into it, I was planning on testing it to make some ETN. While reviewing my notes and the forums, I came accross this thread on SM that linked
to this video https://www.bitchute.com/video/XrjpCnYs7fVL/
He mentioned in the video that he used the same method to make PETN out of ammonium nitrate. It was something that I was looking for as well.
Obviously in the video he doesn't show the purification and recrystalization (but I already know how to do that), and I do have strong reservations
about him simply dumping the whole amount of erythritol into the nitrating mixture, but if it is as good as he says it is (and I will add the stuff
slowly) I am thinking of following those steps.
Except instead of letting the whole thing be at -18C, I'll start at a warmer temperature for PETN. I remember the problems I had with my first
synthesis where my yield was extremely low.
|
|
|
DennyDevHE77
Hazard to Others
Posts: 168
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
The nitration reaction of ETN (at least when ammonium nitrate is used), proceeds much more calmly than the nitration of PETN. I have many times added
tens of grams of erythritol to the nitrating mixture at once, when cooling with cold water, moreover, periodic lowering of the beaker with nitrating
mixture into ice water was enough for effective cooling during the whole nitration (usually 2 hours). Besides, with ETN there is no difference between
+5°C, +10°C, +25°C. And even above +30°C it is hard to lose control, the mixture starts to bubble a bit, at which point you can stir it vigorously
and cool it down.
PETN is quite different. Its nitration is much more active and the temperature can rise much higher. Usually when loading 100g of pentaerythritol it
should be added 1-2 grams at a time, preferably each new portion should be added after adding and dissolving the previous one and stabilizing the
temperature. Ah yes, in sulfur-nitrogen mixture PE dissolves much slower than erythritol. PETN when nitrated with 90-97% acid alone is normally
nitrated both at 0°C and at +20°C. When nitrating with sulfuric acid you need to keep temperatures between +8°C and +15°C. If the temperature is
lower, you get PETRIN (heavy oil, I got it at +3°C), if it is higher, you get badly exploding, yellow PETN (I posted it here once). However, PETRIN
is removed by holding at +30°C after adding all the PE.
To summarize, if you're nitrating ETN, just throw in tablespoons of erythritol into the nitro mixture, stir lazily, and occasionally watch that it's
not above 25°C. You can just keep a glass of nitro mixture in a bowl of cold water. It's with nitrate! If with nitric acid, the reaction is more
active, but still easily controlled.
With PETN, however, strictly watch the temperature. Ice cubes with water will give you +11 to +14. Then soak at +30, then soak for 20 min at +50°C.
Since it is winter, you can use 3 weight parts of snow and 1 weight part of ice, this will give you about -20. Then pentaerythritol can be added in
tens of grams, but again, pentaerythritol takes a long time to dissolve, so after adding all the pentaerythritol, allow a soak of 10 min, stirring it
before moving on to the next steps.
|
|
|
ManyInterests
National Hazard
Posts: 968
Registered: 19-5-2019
Member Is Offline
|
|
| Quote: Originally posted by DennyDevHE77 | The nitration reaction of ETN (at least when ammonium nitrate is used), proceeds much more calmly than the nitration of PETN. I have many times added
tens of grams of erythritol to the nitrating mixture at once, when cooling with cold water, moreover, periodic lowering of the beaker with nitrating
mixture into ice water was enough for effective cooling during the whole nitration (usually 2 hours). Besides, with ETN there is no difference between
+5°C, +10°C, +25°C. And even above +30°C it is hard to lose control, the mixture starts to bubble a bit, at which point you can stir it vigorously
and cool it down.
PETN is quite different. Its nitration is much more active and the temperature can rise much higher. Usually when loading 100g of pentaerythritol it
should be added 1-2 grams at a time, preferably each new portion should be added after adding and dissolving the previous one and stabilizing the
temperature. Ah yes, in sulfur-nitrogen mixture PE dissolves much slower than erythritol. PETN when nitrated with 90-97% acid alone is normally
nitrated both at 0°C and at +20°C. When nitrating with sulfuric acid you need to keep temperatures between +8°C and +15°C. If the temperature is
lower, you get PETRIN (heavy oil, I got it at +3°C), if it is higher, you get badly exploding, yellow PETN (I posted it here once). However, PETRIN
is removed by holding at +30°C after adding all the PE.
To summarize, if you're nitrating ETN, just throw in tablespoons of erythritol into the nitro mixture, stir lazily, and occasionally watch that it's
not above 25°C. You can just keep a glass of nitro mixture in a bowl of cold water. It's with nitrate! If with nitric acid, the reaction is more
active, but still easily controlled.
With PETN, however, strictly watch the temperature. Ice cubes with water will give you +11 to +14. Then soak at +30, then soak for 20 min at +50°C.
Since it is winter, you can use 3 weight parts of snow and 1 weight part of ice, this will give you about -20. Then pentaerythritol can be added in
tens of grams, but again, pentaerythritol takes a long time to dissolve, so after adding all the pentaerythritol, allow a soak of 10 min, stirring it
before moving on to the next steps. |
I suspected that PETN would be hotter than ETN. ETN synthesis is quite mild.
But I am assuming that this nitrate salt method will work and hopefully get good yields. I am getting a little fatigued of basic reagent synthesis.
Ok, so start at -20C for PETN, but it will need a heating step to allow to turn everything into quality PETN and and such. I think we thought we
needed to start at a hotter temperature? I don't got much time now, so I will leave this thought for now...
[Edited on 16-1-2024 by ManyInterests]
|
|
|
ManyInterests
National Hazard
Posts: 968
Registered: 19-5-2019
Member Is Offline
|
|
OK, I am kicking up this thread because I am going to get back into making PETN. The one mistake I did in the one and only time that I made PETN is
the fact I didn't properly control for heat, and I had around 90% HNO3 and I still added sulfuric acid to the mix even when it wasn't necessary.
Also in the heating step, I saw some slight redness come out, I thought it was a runaway and I dumped it in water. I realize that this was probably
not a runaway, as during purifying my RDX in 70% HNO3 I also saw some of those red fumes, but it was clearly not a runaway.
So I still have the old PETN sitting pretty and I am wondering: Does it need the same acid purification that I gave the RDX I had or is the one
acetone recrystalization I gave it sufficient?
After doing the ETN in the ammonium nitrate salt method, I decided that I don't want to do the PETN with that method, since the yields were not nearly
as good as I had anticipated. So I will go the extra effort in making the HNO3 for the whole thing. This wouldn't be that much of a problem, but
getting erythritol is easy, getting pentaerythritol requires me to buy it from an overseas supplier and it is much more expensive.
|
|
|
dettoo456
Hazard to Others
Posts: 258
Registered: 12-9-2021
Member Is Offline
|
|
PETN is most easily prepared via straight WFNA & the polyol itself. Vigorous stirring, around 0-7C, and slow, constant addition rates are key. An
excess of WFNA of at least 2 moles is the lowest I’ve used, and even then, the mixture was very thick and annoying to keep stirred. I haven’t
tried adding a solvent like DCM before though it may be possible (don’t let a DCM/WFNA mix runaway though, it might just combust).
The WFNA I used was slightly yellow and never caused any issues - the rxn mixture would always end up an opaque white by the end of the additions. I
did have a runaway during my last prep due to the mix being too thick and causing localized heating in the mixture, but I still recovered a decent
amount (61% yield) and the product performed within standards.
ETN is far more susceptible to hydrolysis (along with the linear nitrate esters) than PETN or something like TMETN. A thorough washing is still
helpful though. To test, just freshly precipitate some in Acetone or EtOH and use a pH strip on the water/PETN slurry.
If you need pentaerythritol, I’d be happy to assist in procurement - if you’re in the US that is. It seems easy to get in the EU still, and I know
of a Russian supplier still carrying it as well.
|
|
|
ManyInterests
National Hazard
Posts: 968
Registered: 19-5-2019
Member Is Offline
|
|
I live in Canada. It isn't restricted here, it's just that I only know of one seller. I have a little over one KG of the stuff, so I guess I am kinda
exaggerating my fears here. It is a 98% mono, though, and not the 99.6% that some use. I don't know of any place to get 99.6%. But 98% works fine. At
least it did in the first time I tried it, but it has been so long that my next attempt might as well be my first.
What I am going to do is to make sure that I get WFNA. I will distill some nitric acid and instead of relying 100% on specific gravity (which I was
told is often inaccurate), I will just see how it reacts when I blow into it and to see if it burns a hole/sets on fire nitrile gloves. I guess that
would be sufficient to see if it is WFNA?
if I am not satisfied, I will redistill it with a 1:1 ratio of sulfuric acid. Some said I should use more, but nurdrage did it this way, so I'll go
with that.
But just to reiterate my question: Would my first batch of PETN be good as is, or do I need to dissolve it a second time in nitric acid and heat it up
since I mistook the purification for a runaway?
|
|
|
dettoo456
Hazard to Others
Posts: 258
Registered: 12-9-2021
Member Is Offline
|
|
Monopenta at 98% purity is fine. And the mono, di, and tri-nitrate esters of pentaerythritol are soluble enough in water and dilute acids, so that
washing of the precipitated PETN is more than enough to produce pure PETN. Monopenta itself also isn’t soluble in Acetone (and badly in EtOH), so
it’d be removed via recryst.
1kg is more than enough IMO, since PETN should be reserved for caps, and ETN for main charge mixes & plastics.
Density is a less reliable method to find conc%, but as long as you rely on an excess of acid, it’d be fine. Also, if your acid fumes at 0C or
lower, it’s most likely concentrated enough - blowing air on it shouldn’t be necessary. Though bubbling ozone or air through it can help to scrub
NOx.
Magpie’s PPA method (for oleum prep), might also be useful for EM synth. It’d be stronger than Oleum & Ac2O in terms of dehydrating strength.
PPA is solid at 0C - RT.
|
|
|
ManyInterests
National Hazard
Posts: 968
Registered: 19-5-2019
Member Is Offline
|
|
That is good that the 98% is good enough, so I am not worried anymore.
| Quote: | | 1kg is more than enough IMO, since PETN should be reserved for caps, and ETN for main charge mixes & plastics. |
I thought it was the opposite? PETN, especially plasticized PETN is a bit superior to ETN, no? Can you elaborate your reasoning for me please?
Also one thing I noticed with the first PETN batch I made a while go is that there is very slight yellowing, I assume that there was still some acid
left despite one crystalization. I assume this can be remedied with a second recrystalization in hot acetone with more acid scrubbers, like a touch of
sodium hydroxide in the acetone, and crashed in urea-water solution, correct?
I made a 65:35 mix with that current PETN to make an ETN: PETN mix that I plan on loading into blasting caps. From my understanding is that this
mixture will be an extremely brisant mixture that should equal or slightly surpass RDX and as such is a very good. I hope the slight yellowing isn't
going to impact it much. But I do want to salvage my PETN and purify it from the slight degradation it went through.
The experience with that reminds me of the first ever batches of ETN that I made, I noticed they did start to yellow when I loaded from that batch
into caps. This hasn't happened since because I have not only washed my ETN far more, but I also triple recrystalize them. Once in acetone and twice
in methanol, along with adding a good deal of urea and some sodium carbonate/bicarbonate in the crash water.
[Edited on 22-9-2024 by ManyInterests]
|
|
|
dettoo456
Hazard to Others
Posts: 258
Registered: 12-9-2021
Member Is Offline
|
|
Yellowing is always a ‘bad’ sign, but it can be mostly fixed via recryst like you mentioned. I wouldn’t use NaOH though, as it is simply too
strong as a base. Very dilute ammonia solution or a NaHCO3 solution seems better to me. Urea is only meant to be used as a NOx scavenger in storage,
and Ethyl Centralite, Guaiacol, or Syringol will work in the same way as urea, but even better at NOx reduction, as they have more sites to
nitrosate/nitrate.
I’d use ETN in PBX comps more simply due to its lower price, higher OB% (to counteract the plasticizer’s OB%), and it’s manageable sensitivity
when plasticized. It will need to be stabilized, and Guaiacol or Ethyl Centralite at around 0.5-1 W/W% will suffice. The produced plastic should still
be stored away from air, UV light, heat, and not physically manipulated (molding it too much) until use.
PETN is the superior energetic simply from a safety and general performance standpoint, but if you don’t have lots to spare, it’s better suited
for cap applications IMO. A more reliable and safe cap is more important than a reliable and safe plastic - especially if you need the cap to initiate
the PBX.
|
|
|
ManyInterests
National Hazard
Posts: 968
Registered: 19-5-2019
Member Is Offline
|
|
OK. No sodium hydroxide. After dissolving it in hot acetone I will put sodium bicarbonate in the solution along with a few drops of 3% ammonia
solution.
After that I will crash into cold water with urea and more sodium bicarbonate dissolved.
And I see your meaning with why you choose PETN vs. ETN in terms of initiating vs. Main charges. But as I said, will experiment with a 65: 35
mixture. If it proves to be superior than PETN alone I will go with it.
I got sooooo many caps to try out! Look forward to them in the det cap strategy page. Brass, steel, and aluminum bodies, both bottomless and with a
little aluminum plug.
|
|
|
| Pages:
1
..
4
5
6
7 |








