| Pages:
1
2
3
4
5
6
..
25 |
Engager
Hazard to Others

Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
| Quote: | Originally posted by The_Davster
HT-1 is very similar to another energetic. |
Yes i know this one, i asked for this article in References, section. Thiele work with isocyanogen is also avialable there by my request. I have
description of manufacture 1,3-dichloro-2-nitro-2-azoprapane from US patent 4,085,123 witch is attached to the message. But unfortunately i haven't
got acetic anhydride and high vacuum apparatus to preform such preparation. What's why i'm looking for simplier ways...
[Edited on 8-10-2007 by Engager]

|
|
|
franklyn
International Hazard
Posts: 3026
Registered: 30-5-2006
Location: Da Big Apple
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Engager
Isocyanogen tetrazide easily made from isocyanogen tetrabromide and solution of NaN3 in acetone/water |
2,4,6-trichlorotriazine ( Cyanuric trichloride ) similarly serves as the precursor for
2,4,6-triazidotriazine , also made by metathetic action with NaN3. ( See page 432
Chemistry of Powder & Explosives by T. L. Davis.
2,4,6-triazidoborazine , sensitive to impact and friction , one of the very few known
metastable boron compounds ( all azides ) is obtained from a 1 : 3 molar blend of
trichloroborazine and trimethylsilylazide ( Me3SiN3 )
http://www.sciencemadness.org/talk/viewthread.php?tid=1987&a...
the resulting trimethylsilylchloride evaporates from the product.
.
[Edited on 14-2-2008 by franklyn]
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Here is synthesis of 2 copper-nitrotetrazole complexes, both are efficent primaries, and use soluble nitrotetrazole
salts as precursors. This salts can easily be made dissolving acid cooper nitrotetrazole salt (made as described in posts above)in corresponding
hydrohide, heating the solution and filtering off copper oxide. By weighting copper oxide, quantity of nitrotetrazole anion in solution is determined,
this reqired to be known before any other transformations.
Synthesis of precursor nitrotetrazole salts:
Sodium nitrotetrazolate required for first complex is obtained by dissolving acid copper nitrotetrazolate in NaOH
solution. Ammonium nitrotetrazolate is somethat more difficult to produce because ammonium hyrdoxide can not be used dirrectly (copper forms complexes
with it). So to make ammonium tetrazolate acid copper nitrotetrazole salt is dissolved in barium hydroxide, solution is heated to complete exchange
and destroy Cu(OH)2, black copper oxide is filtered off, and weighted to determine quantity of nitrotetrazolate ion in solution. Then ammonium
sulphate is added in stechiometrical ammount to initial barium hydroxide, this results in formation of ammonium nitroterazolate and insoluble barium
sulphate, while excess of barium hydroxide is also destroyed to form some ammonia and barrium sulphate. Solution is filtered from BaSO4 and boiled to
remove residual ammonia, resulting in pure NH4NT solution.
Synthesis of mono-(5-nitro-H-tetrazolato-N)triammine copper(II) perchlorate (MNCuP):
Prepare solutions 10g copper nitrate trihydrate (0.041 mol) in 50 ml of warm water, and 24g (0.25 mol) ammonium
carbonate in 24 ml of 25% ammonia + 30 ml water. Solution of ammonium carbonate is added with stirring to solution of copper nitrate, resulting in
intense blue/violet solution of copper-ammonia complex. Solution is placed on water bath and heated until thin light blue layer of copper basic
carbonate (Cu(OH)2*CuCO3) begin to form at the bottom of reaction flask. During heating period 5g more ammonium carbonate is added in installments.
Solution is filtered from any solid and then 3-4 volumes of alcohol is added. Complex compound is almost insoluble in alcohol and precipitates, solid
is filtered off, washed with alcohol and dried. Product is blue/violet crystalls of [Cu(NH3)3*NH4CO3](NO3). Yield is 75-80%.
To solution of 40 ml 70% HClO4 in 100 ml of water, 3.5g (0.01379 mol) of carbonato triammine copper(II) nitrate in
30 ml of water is added with stirring. Deep blue color of copper carbonate complex quickly turns to transparent sky blue. Resulted mixture is heated
to 60 ± 10С and left to stand at this temperature for half of hour and heated to ~80C, then concentrated solution of sodium 5-nitrotetrazolate
(0.00689 mol) is added by drops with stirring. Mixture is left to sit in boiling water bath for 4 hours, and cooled to room temperature. Product
precipitates as sky blue solid, it is filtered off, washed with small amount of ice cold water and dried. Yield is about 85% of theory.
Photos of products are shown below, left one is carbonato triammine copper(II) nitrate ; right one mono-(5-nitro-H-tetrazolato-N)triamminecopper(II)
perchlorate (MNCuP):
Reaction scheme:
Synthesis of ammonium diaquatetrakis (5-nitro-1H-tetrazolato-N2) cuprate (II) (NH4CuNT):
Solution of 5.5g ammonium 5-nitrotetrazolate (made by chain: CuNT => BaNT => NH4NT) in 38 ml of water added with stiring to solution of 2.52g
Cu(NO3)2*6H2O in 110 ml of water. A small quantity of blue precipitate is formed emidately. Solution was heated on boiling waterbath for 4 hours,
solution becames transparent blue. It's slowly cooled to room temperature and after to 10C in freezer. Large quantity of blue "snowy" precipitate is
formed, solid is filtered off, washed with ice cold water and with small portion of ice cold alcohol. Product was air dried.
Photo of product shown below:
Reaction scheme:
Properties of complexes:
MNCuP: [Cu(NH3)3(NT)](ClO4) - sky blue solid with bulk denstity about 0.65, irregulare plate structure. Sensitive to impact and friction but less then
conventional primaries (similar to BNCP). Digital data about explosive properties is unavialable now, and will be investigated by later research.
NH4CuNT: (NH4)2[Cu(NT)4(H2O)2] According to patent data, density is 1.94 g/cm3, detonation velocity 7390 m/sec (at
1.71 g/cm3). Insensitive to spark up to 0.36 J (human activity generates up to 0.25 J), sentive to shock 23 cm (vs 9.6 PbN3 and 14 PETN), slightly
sensitive to friction 0.6 kg (vs 0.01 PbN3 and 5.8 for PETN). Thermicaly stable up to 265C, detonation products volume is about 750 l/kg, products of
explosion: N2, CO2, H2O, ~2% NO2, ~3%CO. Oxygen ballance (CO) is zero. Substance is stable on air, light and moisture. Almost completely safe then
wet, even with open flame. In dry state flame contact takes DDT (deflagration-detonation transition).
I've also tried Fe and Co complexes. Attempt with FeCl2*6H2O was unsuccesfull, as i've mentioned in previous post. Attempt with Co(ClO4)2*6H2O gave
success but yield was low. I guess there are some special conditions that need to be satisfied then making Fe and Co complex 5-nitrotetrazolates.
[Edited on 30-10-2007 by Engager]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
guanylazide 5,5'-Azotetrazolate (possible?)
| Quote: | Originally posted by Engager
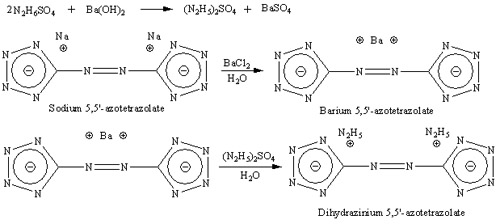
Synthesis of dihydrazinium 5,5'-Azotetrazolate (HZT)
Prepare solution of 4.8g sodium 5,5'-azotetrazolate in 30 ml boiling water and 5.58g barium chloride dihydrate in 15 ml of boiling water. Solutions
are mixed and stirred, precipitate of barium 5,5'-azotetrazolate forms emidately, solution is cooled to 10C and filtered. Ba salt is washed with small
amount of ice cold water and dried at room temperature. Yield is about 6.2g.
Make solution of 5.3g hydrazine sulphate (N2H6SO4) in 155 ml of water, 6.44g of barium hydroxide is added with stirring, and after 6.2g of barium
5,5'-azotetrazolate is added. Mixture is well stirred for 1 hour, solid (BaSO4) is filtered off and discarded, resulting in yellow solution of
dihyrazinium 5,5'-azotetrazolate. Solution of HZT is placed on boiling water bath and heated until most of water evaporate and first crystals of HZT
form. Solution is then removed from water bath and cooled to room temperature and after in freezer to 10C. Mixture almost comepletely solidifies to
form yellow needles of dihydrazinium 5,5'-azotetrazolate dihydrate. Crystalls are filtered of and dried at room temperature. Yield is about 87%.
Anhydrous salt may be obtained by drying dihydrate in vacuum exicator at 100C for 2 days.
I've already made dihydrazinium azotetrazolate. I can post metod of synthesis with photos if somebody interested. It's yellow needle like solid,
soluble in water. Photos shown below, left photo is barium azotetrazolate, two others are dihydrazinium azotetrazolete (HZT):
  
Reaction scheme:
[Edited on 14-9-2007 by Engager] |
(reaction scheme stored on local server)
Sorry for not mentioning this earlier ....
It would seem that the guanylazide salt would have more interest in terms of an energetic material and could be made
by a parallel reaction , using (mono)guanylazide sulfate (exist?)
substituted for the dihydrazine sulfate , the parallel
reaction expected to follow the course of your last reaction shown above .
The predicted product would be
guanylazide 5,5'-Azotetrazolate .
The possible usefulness of guanylazide ,
and (though less convenient) nitroguanylazide ,
as well as triaminoguanidine , should not be overlooked
as possible "metal substitutes" in the formation of energetic salts derived from the various energetic acid tetrazoles .
I don't know of any references concerning such materials ,
so extreme caution would be prudent in experiments concerning such possible compounds .
P.S. As Axt pointed out earlier , you should upload , copy your attachments into the forum ftp , so they will not become dead links and empty space ,
(lost data) later , but are safely archived and backed up here .
[Edited on 30-10-2007 by Rosco Bodine]
|
|
|
quicksilver
International Hazard
Posts: 1820
Registered: 7-9-2005
Location: Inches from the keyboard....
Member Is Offline
Mood: ~-=SWINGS=-~
|
|
| Quote: | Originally posted by Rosco Bodine
Yeah it is in PATR under " Tetrazene " , an Australian
government study found that stab detonators containing
tetracene would fail to function after storage at elevated temperatures because of the tetracene slowly changing to
5-aminotetrazole by thermal decomposition . They stated
complete conversion at 90C in less than 3 days .
|
Does anyone know how the decomposition mechanism works within this chemical? I have seen this personally in that after a period of weeks the tetrazene
produced via the COPAE would no longer produce the black-smoke deflagation it had previously. But in this case the exposure level was perhaps 40 C
over the course of weeks or months. Is tetrazene unstable @ room temp?
An addendum: it appears that 40C is not a contributor to instability but 30+ degree temp fluctuation may be. The only accountable issue would be that
or UV from a extremely low level source.
[Edited on 14-2-2008 by quicksilver]
|
|
|
The_Davster
A pnictogen
Posts: 2861
Registered: 18-11-2003
Member Is Offline
Mood: .
|
|
synthesis of azidotetrazole from cyanogen bromide:
(as requested)
Attachment: cyanogen azide.pdf (640kB)
This file has been downloaded 2342 times
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Today i've made batch of acid copper nitrotetrazolate for fourth time. Unlike my previous experieces, this time i have encountered "minor detonations"
during same procedure as i've used before. These microdetonations are caused by nitrogen oxide fumes from the reaction solution reacting with
droplets of 5-AT on surfaces in the reactor to form 5-diazotetrazole which may spontaneously detonate in
solution when the concentration exceeds 1%. Aside from being psychologically disturbing, these micro-detonations may be strong enough
to break glass and may result in release of the potentially explosive reaction mixture. Detonations are accompanied by white flashes and loud report,
you got to have a steel nerves to stay not worried about such scary stuff, especialy then mixture is allmost full of precipitated acid copper
5-nitrotetrazole salt.
Reasons why this time they arrisen remain unclear, they seem to appear randomly, without any correlation with reaction state. However i've found that
in round bottom flask, they appear much more frequently, then in flat bottom flask, and also depend from flask volume and neck width. This may be
connected with temperature gradients and vent efficency inside reaction wessel.
Temperature also plays a role, surprisingly, when it is low microdetonation probability rises greatly, so it is very important to keep temperature
between 15 and 18C at ALL points of rection mixture. This may be acchived by adjusting reagent additon rate and by effective stirring. Mixing rod edge
must be round and smooth to produce minimal impact if it accidentaly hits the bottom during stirring. At low temperature, glass stiring rod with
relatively sharp edge, causes minor detonations at almost any accidental contact with flask bottom.
So there are some additional notes to procedure to avoid minor detonation problem. Reaction wessel must be flat bottom flask with large volume and
wide neck, stirring rod should not have sharp or low area bottom edge to maximize impact area. Additon rate must be slower at least twice then 90 mins
mentioned in literature, addition shoud be not more then 0.5 ml at one time, perfectly drop by drop. Efficent stirring and temperature control are
essential, temperature during addition time must be kept strictly between 15 and 18C in all points of reacion mixture.
Don't bother literature souces say that minor detonations are relatively safe, whey must be taken very serious!!! They still can do nasty dammage to
glassware, and may be even able to initiate exposion of whole mixture. This synthesis of 5-nitotetrazole must be taken as hazardly dangerous, and must be carried out only by experienced chemist, with all necessary safety measures.
[Edited on 21-2-2008 by Engager]
|
|
|
The_Davster
A pnictogen
Posts: 2861
Registered: 18-11-2003
Member Is Offline
Mood: .
|
|
Interesting to hear of your detonations, I have prepared the copper salt in small round bottom or erlenmeyer flasks with magnetic stirring 3 times
now, and have never experianced this. Good to know it can happen even with copper present in the ATZ solution being added. I believe due to a too
slow addition I went to 10C once without experiancing these microdetonations.
Back to info on the tetrazylazide as discussed on the first page of this thread:


|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Russian references as i promised, sorry man, too large to attach, i had to place them on Rapidshare.
http://rapidshare.com/files/97898324/Davster.rar.html
|
|
|
oket
Harmless
Posts: 1
Registered: 7-2-2008
Member Is Offline
Mood: No Mood
|
|
"SOME REACTIONS OF THE AZOTETRAZOLE ANION
WITH DILUTE MINERAL ACIDS"
5-Hydrasinotetrazole
To a solution of disodium asotestrazole pentahdrate (10.0 g) Tuspended in
water (100 ml) was added hydrochloric acid (25 m1 5N). The solution was
warmed on a water bath until the gas evolution finished, then was evaporated
to dryness from water three times to remove the hydrochloric acid. The
residue was dissolved in a minimum of hot water and a hot solution of sodium
acetate (10.0 g) in water (10 ml) was added. The apparatus was flushed out
with carbon dioxide and the solution allowed to cool to give 5-hydrasinotetrasole
as white prisms with a yield of 2.5 g# mp 195 - 8°0 (lit 199d).
Attachment: SOME REACTIONS OF.pdf (1.3MB)
This file has been downloaded 1632 times
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Bis-(5-nitrotetrazolato-N2) cobalt (III) perchlorate (BNCP).
Orange-red needle-shaped crystalls insoluble in cold water. Density 2.05 g/cm3, thermicaly stable below 200C, decomposes at 269C. Heat of explosion
3.32 MJ/kg, detonation velocity 8100 m/sec (at 1.97 g/cm3), deflagration - detonation transition takes about 10-11 microseconds, minimal intiating
charge vs RDX is 0.05g. Non toxic, not hygroscopic, sensitive to fire, relatively insensitive to impact and friction (on secondary explosive level).
Gives 50% explosions in drop test with 2.5 kg weight and 17 cm drop altitude (PETN - 12 cm), insensitive to electrostatic discharge. Ignite from Nd
laser beam (wavelenth 1.06 micrometers, impulse time 1 msec, beam diameter 0.5 mm, impulse energy ~ 1.5J). Can be used as primary in detonators
without secondary - bosster charge. Has optimal density interval, if density is too low burns - without detonation, if too high - becomes overpressed.
Used in safe electric detonators and futuristic light ignition detonator systems.
Synthesis of BNCP
1. Preparation of [Co(NH3)4CO3]2SO4*3H2O complex. 47g CoSO4*7H2O is dissolved it 125 ml of water and is added to
solution of 100g (NH4)2CO3 in 500 ml H2O + 250 ml 25% ammonia solution. Resulted dark violet solution is oxidised by addition of 14 ml of 30% hydrogen
peroxide. Solution is allowed to sit for 30 minutes, after that period it is placed on boiling water bath and is evaporated to 300 ml volume. During
course of the concentration process, solid ammonium carbonate (25 g) was added in installments. Solution is filtered from unsignificant ammount of
precipitated black cobalt oxide and is further concentrated to 200 ml volume. Solution is slowly cooled to room temperature, product precipitates as
small deep-red prisms. They are filtered out from the solution and allowed to dry at room temperature. Yield is 16 g of pure product, mother liquer
can be further concentated with addition of ammonium carbonate to get more 16 g of complex, but it is less pure.
2. Preparation of [Co(NH3)4CO3](ClO4) complex. 16g of complexed synthesised on first stege is dissolved in 320 ml of
water. Solution is filtered and 16g of sodium perchlorate in 40 ml of water is added with stirring. Mixture is cooled on ice (or in freezer) for 3-4
hours. Product precipitates as small, lustrous, sharp violet prisms. Crystalls are filtered off, washed with small ammount of ice cold water and dried
at room temperature. Yield is about 14 gramms.
3. Preparation of BNCP. 14g [Co(NH3)4CO3](ClO4) is dissolved in 140 ml 10% perchloric acid, and solution of 26.5g of
sodium 5-nitrotetrazolate dihydrate in 450 ml of water is added with stirring. Solution is placed on boiling water bath, and allowed to sit there for
4 hours. Solution is slowly cooled to room temperature and then cooled in freezer to 10°С, precipitated crystalls of BNCP are filtered of,
washed with cold water, and recrystallised from 1% perchloric acid. Yield is 12.9g.
Photos of reaction products: first is tetraminocarbonato cobalt (III) sulphate trihydrate (product 1), second is tetraminocarbonato cobalt (III)
perchlorate (product 2), third is crystalls of BNCP.
This is the video of deflagration-detonation transition of BNCP in paper tube. Sorry i know i have to attach this file to post, but it seems to bee
too large, i tried 2 times with no result. So i have to place it on rapidshare:
DDT of BNCP video
[Edited on 11-3-2008 by Engager]
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Hello Engager, very very nice! Inorganic syntheses of beautiful compounds, yet with such interesting properties!
Recrystallised from 1% HClO4, is the compound unstable or why is this necessary? Did you base your synth on a published report (if so, pls say) or
work it out yourself? I imagine the former as you also cite a lot of det data.
I was wondering whether you would upload the video to utube, it's likely to last longer there, and at least I can download it there!
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Recrystallization is for purification, it is not necesary but can be done if needed. And i forgot to post reaction schematic:
References are on Russian language, i attached them to the bottom of this post.
[Edited on 12-3-2008 by Engager]
Attachment: BNCP Russian References.rar (188kB)
This file has been downloaded 1704 times
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I would have guessed a different outcome for that last reaction which would essentially be a mixed salt of the
cation tetrammine Co dihydroxide complex , with one nitrotetrazolate anion and one ClO4 anion .
The same reactions should probably occur for copper and probably also for nickel .
I wonder , and have some skepticism  actually , about actually , about
the need for the "carbonato transition" shown . It seems
the entire carbonate intermediate is unnecessary .
The reaction scheme seems so closely parallel to the reaction where tetrammine copper (di)perchlorate is precipitated from the double decomposition
reaction of concentrated copper nitrate solution added to a warm NH4OH solution of NH4ClO4 .
What I see as more likely for Cobalt compound III ,
is that central two hydrogens on the extreme right ,
which are associated with the hydroxyls , are *not*
supposed to be there at all .....but simply the two
hydroxyls which define the dibasic structure of the
tetrammine cobalt "dihydroxide" base . It is a base ,
*not* a dihydrate as shown .
The subcase "3" for the ClO4 group in the last line
should be 2 not 3 as shown .
And the final molecule should have the ClO4 group substituted for the lower nitrotetrazolate inside the
bracketed larger structure , with a subcase of *2*
outside the bracket .
That's the way I see it as more plausible anyway .
And one last thing , it doesn't seem possible that this
substituted tetrammine cobalt perchlorate would be stable in neutral or acidic aqueous solution , but only in an ammoniacal solution of mixed ammonium
nitrotetrazolate
and ammonium perchlorate . In aqueous solutions otherwise it should decompose via hydrolysis , the
same as does tetrammine copper perchlorate .
Being structured and reportedly behaving so differently
from what would be expected , it is certainly anomalous
in many ways if all of this is not true .
[Edited on 12-3-2008 by Rosco Bodine]
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Yea, this all is not my invention, read refs i provided. Cobalt with ammonia forms Co(NH3)6 and NH3 can not be substituted with 5-nitrotetrazole,
carbonate is only a "protective" group, whitch is finaly destroed on final stage of synth to free 2 coordination places for NT. Tetramine complex is
stable in acid even in 30% HClO4 you can make it for yourself to see this is true, so all is correct, read more about cobalt complexes before making
your statements. Your are denying practicaly observed and documentaly patented way of synth, may be you just go on and got top Nobell chemistry award
if you are so clever.
[Edited on 12-3-2008 by Engager]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
ooops !
Pardon my brain fart above . I just went back and reread
the text description and equations and saw the use of H2O2 to change the valency of the Co +II to Co +III in the first reaction which then changes
everything to follow . The +III valency I was simply reading as "compound #3" and not paying attention it was the valency designation for the Co .
My Nobel prize will have to wait until I clean my glasses and have another cup of coffee
Interesting compound , and the reactions do make a lot more sense now .
Edit: I still have my reservations about the hydroxyls .
With a Co+III complex , should not we see a hexammine cobalt trihydroxide as the base ?
[Edited on 12-3-2008 by Rosco Bodine]
|
|
|
hokk
Harmless
Posts: 3
Registered: 28-2-2008
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Engager
Synthesis of BNCP
|
I don't know if these are stupid questions or not, but anyways:
Could I use CoCl2 instead of CoSO4 without any problem? (changing the added mass of course)
Could I also use (NH4)HCO3 instead of (NH4)2CO3 if I add more of the (NH4)HCO3?
Thanks!
[Edited on 20-3-2008 by hokk]
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
No, you can't use CoCl2 because it will form complex with different properties and structure. Synth of Werner complexes is quite delicate and require
good replication of synth procedure. You can use NH4HCO3, but procedure have to be modified. This synth is taken from literature, but if you modify it
to use NH4HCO3 it can work but also may fail because a different solubilities/concentrations, so tryout is needed.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Cobalt nitrate and ammonium carbonate might work okay .
As for substituting the bicarbonate , it seems like that should work okay if you add extra NH4OH
sufficient for conversion of the acid carbonate to the normal carbonate .
Hmmmm....according to this article attached , the chloride
does form the analogous carbonatotetrammine cobalt (III) chloride ,
as part of a series of analogous salts derived
from different cobalt salts used as the starting material .
The efficiency for conversion to the perchlorate would be solubility related .
But it doesn't appear structure would be any issue .
Engager is likely incorrect about the chloride not being workable .
[Edited on 20-3-2008 by Rosco Bodine]
Attachment: Carbonatotetrammine cobalt (III) nitrate.pdf (91kB)
This file has been downloaded 1945 times
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
carbonatotetrammine cobalt (III) chloride
There is a preparation of the chloride analogue
described in the attached article where it is an
intermediate used in further synthesis .
Attachment: carbonatotetrammine cobalt (III) chloride intermediate.pdf (124kB)
This file has been downloaded 2210 times
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Recently i've got interesting idea how bis-tetrazole can be made without use of dicyan as starting product. I found rection in Elderfield book of
heterocyclic chemistry, there was a reaction of HN3 with oximes, it yelded corresponding tetrazole, so i thought to use this scheme to generate
bis-tetrazole from glyoxime. Starting products will be glyoxal and hydroxylamine, they condense readily to form glyoxime, whitch is then reacted with
2 moles of HN3 to yield 5,5'-bistetrazole:
However i have not found this synth in literature, but i think it can work. Literature methods use Mn to precipitate insoluble Mn bis-tetrazole salt,
but case above this can not be made because one don't want to allow Mn azide to form. Excess of azide can be destroyed by using acidic nitrite
solution on boiling, this will destroy azide: HN3 + HNO3 = (boiling) => N2 + N2O + H2O
Also there is one problem - low solubility of glyoxime in cold water, this forces to preform reaction in hot solution - this will generate a lot of
fumes of volatile hydrogen azide, what is dangerous.
Somobody got any ideas about this? Will this synth work, and how to rise solubility of glyoxime in cold water?
[Edited on 23-3-2008 by Engager]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
convergence of earlier posts
| Quote: | Originally posted by Engager
I've also made two other tetrazole-based energetic compounds. Diazoaminotetrazole and dihydrazinium 5,5'-azotetrazolate (mentioned as substance with
highest known positive heat of formation).
Synthesis of sodium bis-5,5'-diazoamintotetrazolate from aminoguanidine bicarbonate
Prepare mixture of 11.5 ml 70% nitric acid with 100 ml of water, add by portions with stirring 24.8g aminoguanidine bicarbonate. Stir mixture until
CO2 evolution stops and then add 20.4 ml 70% acetic acid. Mixture is stirred and slightly heated until all solid dissolves. The resulting clear
yellow solution is solution of aminoguanidine nitrate in 12-13% acetic acid. This solution is cooled in freezer to 3-4C, well mixed and placed on ice
bath. Slowly, with stirring, by small portions at time ice cold solution of 17.5g sodium nitrite in 75 ml of water is added. While addition,
temperature must be all times kept below 12C, perfectly in interval of 5-7C, process takes about 30-40 minutes. After diazotation is finished mixture
is removed from ice bath and left to stand at room temperature for 24 hours. Some time after removal from ice bath slow evolution of nitrogen begings,
and mixture can heat up to 25C, this is ok, so don't worry, and after about 12-16 hours of standing evolution of nitrogen stops and large amount of
diazoaminotetrazole precipitates. Solid is filtered off, washed with ice cold water slightly acidified with acetic acid and left to dry at room
temperature. Yield is about 50% of pure mono-sodium salt of diazoaminotetrazole. Photos of product shown below:
Reaction scheme:
[Edited on 11-9-2007 by Engager] |
On page 2 of this thread , in the second post there is a similar synthesis reported in US2064817
which uses a sodium acetate - acetic acid buffer for the diazotization .
I wonder if the buffer system has been tried and compared
with your described method , to see if the use of the buffer
can improve the yield above 50% . Also , even though
it has been specified that a non-mineral acid buffer system
be used , I wonder if boric acid - sodium borate has ever been tried and found to be unworkable .
Related to your interest in the metal ammonia complexes of these tetrazoles ,
which I have been following with interest here , also on page 2 of this thread was reported a
Copper Ammonium Salt of Diazoaminotetrazole , described
in US2004719 . On page three you show a sample of the
non-complexed parent compound in matchbox #3 which
you called CuDAT .
| Quote: | Originally posted by Engager
I've attached photo of silver 5-nitrotetrazolate, among other tetrazole derivatives i've made so far. Upper-left to lower-right: 1. Copper-ammonium
complex 5-nitrotetrazolate (NH4)2[Cu(N4C-NO2)4(H2O)2] - green primary (NH4CuNT) ; 2. (N2H4)2(N4C-N=N-CN4) - dihydrazinium 5,5'-azotetrazolate (HZT) ;
3. Cu3(N4C-N=N-NH-CN4)2 - copper 5,5' diazoaminotetrazolate (CuDAT) ; 4. Cu(N4C-NO2)2*(HN4C-NO2) - acid copper salt of 5-nitrotetrazole (CuNT) ; 5.
Ag(N4C-NO2) - silver 5-nitrotetrazolate (AgNT) ; 6. Na3(N4C-N=N-NH-CN4) - sodium 5,5'-diazoaminotetrazolate (NaDAT) ; 7. Na2(N4C-N=N-CN4) - sodium
5,5'-azotetrazolate (NaAT) ; 8. HN4C-NH2*H2O - 5-aminotetrazole monohydrate (ATZ).
[img]http://www.sciencemadness.org/talk/viewthread.php?action=attachment&tid=8144&pid=106891[/img] |
Your picture of CuDAT is not clear enough to be sure , but looks to be a crystalline lump material ,
whereas the patent US2004719 describes the CuDAT as being an amorphous powder .
I am curious about your method of synthesis for the CuDAT , and also am curious if you tried the complexing
with ammonia as described in US2004719 . There is only
mention of the usefulness of the ammonia complex as a
sensitizer component in priming mixtures where it is used as a small percentage .
Is there more information concerning the possible usefulness of the complex alone ,
or the parent compound CuDAT as an initiator ,
described in the Russian literature ? Have you done any experiments with either
the CuDAT or its diammine ammonia complex to see if these
compounds have usefulness as "green primaries" ?
[Edited on 23-3-2008 by Rosco Bodine]
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Stickers on boxes may not be correct subtance with sticker Cu(DATZ)2 on box is copper diazoaminotetrazole, that's why i listed correct names above
picture. Actualy Cu diazoaminotetrazole is amorphous, but when it dried it stucked to paper in some kind of lumps, in wet state it's olive green, but
dry it is almost black. It's quite sensitive to friction, it exploded then i tried to grind it in mortar. Explosion was quite loud, but i don't find
any usefull applications for it in literature, only mentions about it's existance. Surely it is highly sensitive and powerfull explosive, but i'm not
sure about it's initiative power. I have not tried ammonium complex, because i think it's too sensitive.
I attached photo of wet product below:
[Edited on 23-3-2008 by Engager]
.jpg - 58kB")
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I was thinking that possibly the CuDAT or its diammine complex might substitute for lead azide
in a binary composition with barium styphnate , as a lead free primary . This binary is supposed
to have a greatly reduced mechanical sensitivity , excellent stability , good initiating power
and is cheaply made .
See US3284255
http://www.sciencemadness.org/talk/viewthread.php?action=att...
Another possible binary would be a mixture of the CuDAT or its diammine with guanylazide picrate .
These same binary mixture schemes might also be useful with some of the other
complexes which have been described , as a means of improving the economics
and lowering the sensitivity for the combined mixture .
Another possibly useful guanylazide is the styphnate ,
but I have found no mention of this in the english language literature .
Is there any mention of guanylazide styphnate
in the Russian literature ?
[Edited on 23-3-2008 by Rosco Bodine]
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
I've tried synth of bis-tetrazole from previous post, with no success. I've used following procedure: 10.7g of sodium azide and 13g of ammonium
chloride are dissolved in 150 ml of water, then 8.8g of glyoxime was added. Mixture is heated on water bath, until glyoxime fully dissolved and then
refluxed for 6 hours. No collor change occured, no glyoxime precipitated after cooling, then 16.8g of soda is added to destroy ammonium salts, and
solution is boiled on water bath for more 2 hours. Solution color changed from pale yellow to deep red, no product precipitated after cooling.
Unreacted azide is destroyed by action of nitrous acid: 25 ml of sodium nitrite is dissloved in 100 ml of water, and is added to red reaction mixture,
then solution of 20 ml conc. sulphuric acid in 140 ml of water is added by small portions. Destruction of azide is accompanied by virgous evolution of
nitrogen oxides, and foaming, addition of sulphuric acid was continued until mixture show acidic pH, iodine-starch paper test shown blue singnal
(means excess of nitrite in reaction mixture). Reaction mixture was allowed to sit for a night, and after very large prismic crystalls are
precipitated - longest one 7 cm lenth with 0.5 cm width. Presence of bis-tetrazole was analyzed by addition of manganeese (II) sulphate (Mn salt of
bis-tetrazole reported to be insoluble in water), no precipitate was obtained from mother liquer and from solution of precipitated crystalls. So
structure of product obtained is unknown, but it is not bis-tetrazole, because no precipitate with MnSO4. Structure of red intermediate in solution
during synth pathway is aslo unknown.
Today i thought another way to make bis-tetrazole without use of dicyan. It's simmilar to synth of 5-aminotetrazole by Thiele method. Glyoxime must
react with hydrazine forming corresponding hydrazone, whitch is "dimeric" analogue of aminoguanidine, diasotation with HNO2 should result in "dimeric"
guanylazide, whitch should form bis-tetrazole in way simmilar for guanylazide=>5-aminotetrazole. Proposed reaction scheme:
Any ideas about this possible path? Sobody have any information about intermediate products and their properties? Well be glad to hear any oppinions
about this synth path. The_Davster! What do you think about it?
[Edited on 28-3-2008 by Engager]
|
|
|
| Pages:
1
2
3
4
5
6
..
25 |
|


























