| Pages:
1
2
3
4 |
DNA
Hazard to Others

Posts: 191
Registered: 11-6-2003
Location: @moon
Member Is Offline
Mood: Experimenting
|
|
I used 300mg substrate, I've noticed that 5% works not as good as 10% Pd/C, although often with catalytic hydrogenations the reaction works better
when trying it again with a little bit less catalyst then doing it again with even more catalyst.
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
today, i've got ~97% from a nitro CTH reduction.
the most important thing is to make sure that the ammonium formate is COMPLETELY dissolved before adding the catalyst. i fitted a lab glove (pinched
with a needle to make a tiny leak) to the top of the reflux condenser so a slight overpressure of H2 was maintained.
actually, i think that the reduction only proceeds if enough H2 is absorbed by the catalyst. thus, it seems much more viable to generate H2 from Metal
or Hydrides and H+ instead of buying totally overpriced ammonium formate (which also increases solvent volume to insane levels (750ml MeOH for 370mmol
substrate).
the reduction should run just as lovely with a balloon fitted to the top of a 3-neck flask.
[Experimental Procedure]
390mmol of 4-Fluorophenyl-2-nitropropane was mixed with 750ml MeOH, 75ml dH2O and 124g (5eq) Ammonium Formate and mechanically stirred until all the
formate was completely dissolved.
12.5g Pd/C (10%) was then added, which caused an exothermic reaction in the course of a few minutes and the MeOH started refluxing.
A chemical disposable glove (pinched with a needle to make a tiny leak) was fitted to the top of the reflux condenser by means of a quickfit
thermometer adapter (nifty, eh?) to maintain a slight overpressure of H2.
After stirring overnight (approx. 14hrs), the catalyst was filtered off and the MeOH was removed on a rotary evaporator.
The distillation residue was acidified and extracted 3 times with 50ml Toluene (which was unnecessary). After basification of the reaction mixture
with 25% aq. NaOH, an amine phase immediately separated.
The basified mixture was extracted with 3x100ml toluene each, and the combined organic extracts attempted to be dried with K2CO3, which failed due to
the formation of an unfilterable gel-like solid.
It is important to note that in the reduction of 4-Fluorophenyl-2-nitropropane, another unfilterable, probably colloidal solid may remain in the
organic extracts, no matter what reduction method is being used. This is most easily removed by distillation of the product.
The toluene was removed on a rotary evaporator, which azeotropically removed the residual water.
The remaining residue of 4-Fluorophenyl-2-aminopropane weighed 58g (378mmol, 96,9% of theory) and was stored under Ar, over KOH in the freezer.
4-Fluorophenyl-2-aminopropane easily combines with CO2 from the atmosphere to form spectacular flower-like crystalline structures.
[Edited on 31-3-2008 by stoichiometric_steve]
|
|
|
DNA
Hazard to Others
Posts: 191
Registered: 11-6-2003
Location: @moon
Member Is Offline
Mood: Experimenting
|
|
Let me give you a tip, presaturate your solutions before adding anything with H2 gas, then after adding your chemicals (after each compound) saturate
with H2 gas, it will improve yields. I haven't got the reference now at hand but will post it asap.
Could you give a bit more details?
Like what substrate did you use?
What volume of solvent?
What solvent (MeOH)?
Rate of addition of the substrate?
Was the substrate added as it is or as a solution or with the aid of a soxlhet extractor?
How long did you let the reaction stirr?
At what temperature did you do the reaction?
Did you monitor with TLC?
[Edited on 31-3-2008 by DNA]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by StoechiometricSteve
actually, i think that the reduction only proceeds if enough H2 is absorbed by the catalyst. thus, it seems much more viable to generate H2 from Metal
or Hydrides and H+ instead of buying totally overpriced ammonium formate (which also increases solvent volume to insane levels (750ml MeOH for 370mmol
substrate).
the reduction should run just as lovely with a balloon fitted to the top of a 3-neck flask. |
Yes, i also though this could be just aswell if not better. At first i was thinking of saturating the catalyst in MeOH with H2, then add the
formate, then i figured i could just aswell use straight H2 for the reduction. But having a H2 saturated MeOH/Pd/c suspension presents a significant
fire hazard when opening if not using argon flow...
Using a formate also present less hazard du to h2 gas, but i find it hard to judge exactly how much h2 is evolved when using k formate. Never
bothered trapping the gas.
For atm hydrogenations, i was thinking of using something like Al + dilute h2so4 or naoh, dripped in, with an inverted cylinder to moniter the h2
uptake, and a drying bottle, a ballon attached onto of the condenser of the reaction flask. So the inverted cylinder could filled with h2 by slowly
adding base/acid to al shreddings, cutting the h2 flow when the cylindrer is full, taking care not to let any pressure build up, and refill the
cylinder by adding more acid/base when needed. Such a device is described in a article of the raspberry ketone thread IIRC.
I've never used any H2 from a metal + acid or base, could someone who has tell me how quick/long lasting the h2 evolution is when portions are
added? The whole inverted cylinder might not be a good idea if it is too continous, overpressure of h2 wouldn't be a good thing...
DNA, have you tried out some more nitrostyrene reductions using atm hydrogenations lately?
|
|
|
DNA
Hazard to Others
Posts: 191
Registered: 11-6-2003
Location: @moon
Member Is Offline
Mood: Experimenting
|
|
Not anymore, I've been doing more LiAlH4 reductions in Et2O and in THF and in a mix of Et2O and THF.
Since shulgin in pihkal uses Et2O for the 3,4,5-trimethoxynitrostyrene substrate and for others he uses THF which I think is weird.
So I tried on 3,4-dimethoxynitrostyrene the reduction with THF and with Et2O and a mix.
With Et2O I could reflux for 48h and still there was 80% precusor left.
With THF reduction was done in 20minutes at RT.
With saturated solution of nitrostyrene in THF and the LiAlH4 in Et2O the reduction went also smoothly in 30 minutes after complete addition.
Will try soon 3,4,5-trimethoxynitrostyren with the aid of a soxhlet extraction apparatus.
Actually I got a bit fed up with the nitroalkane / CTH reduction.
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by DNA
Actually I got a bit fed up with the nitroalkane / CTH reduction. |
you shouldn't be 
i've just tried out the method for the double bond reduction which uses Toluene/Water with a PTC (i used Aliquat 336), and it worked like a charm for
4-Fluorophenyl-2-nitropropene, which is usually a pain in the ass if EtOH/EtOAc solvent systems are used (because the nitronate crashes out and forms
an intractable, gum-like mass that only very slowly dissolves in water - it can break your stirrer).
My guess is that this also works for Nitrostyrenes, since the PTC is supposed to carry inorganic anions (BH4- in this case) to the organic layer, and
not the Nitrostyrene to the water phase where the nitronate resides. However, a lot of people got tarry product when using this method with all the
Nitrostyrene added at once.
I used a Toluene solution of the substrate (1mol would need 450ml of Toluene) which was slowly added to the aqueous NaBH4 solution (~2eq. NaBH4)
containing some NaOH to reduce decomposition.
The workup was incredibly easy...acidify (while cooling with ice!!!), separate the toluene, extract water layer with toluene and
remove toluene. some unreduced substrate made it into the distillation flask and polymerized to a black mass upon heating. nevertheless, the distilled
nitropropane was clean as fnack. i got 98% 
[Edited on 31-3-2008 by stoichiometric_steve]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Nice! Doesn't the PTC cause any problems with the nitro reduction? I would be a little concerned by catalyst poisoining, no?
I take it you haven't tried that on nitrostyrenes yet?
DNA, samsung (sp?) got excellent results using the EtOAc/EtOH NaBH4 reduction of 3,4,5-MeO-nitrostyrene. The nitroethane is reduced in good yields
(>70%) with Pd/C K formate. I heard this substrate doesn't perform that well in LiAlH4 reduction, so I would rather keep this hydride for other
nitrostyrenes. Just an advice.
I'd love to hear more on your LiAlH4 reductions though. Strange to see such difference when using Et2O! I always took it granted that both THF and
diethyl ether were perfectly echangeable in such reduction, although i have only used commercial LiAlH4 solution in THF up to now.
|
|
|
DNA
Hazard to Others
Posts: 191
Registered: 11-6-2003
Location: @moon
Member Is Offline
Mood: Experimenting
|
|
A commercial LiAlH4 solution is indeed safer, I still have sweat on my forhead everytime I add LiAlH4 to the ether or THF...
But well if you use a 1M solution you need liters of LiAlH4 solution if you want to prepare a little bit bigger amount.
So far on the 3,4,5 (starting with 1,7g nitrostyrene) I got a yield of 55% (sulphate salt) while NOT using the soxhlet extractor ( I used not so much
Et2O as shulgin did).
I got a 78% yield on 3,4-diMeOnitrostyrene when using THF in 20 minutes.
In a couple of days I will do the 3,4,5-trimethoxynitrostyrene it will undergo a reduction at 4,8g scale with 4g LiAlH4 in Et2O with the aid of a
soxhlet extractor with Et2O (so actually the exact procedure as in pihkal this time).
But my interest got back when I read that a >90% yield was obtained by stoichiometric_steve I still have quite some grams of 10% Pd/C lying around
and well I did got a 97,5% yield on the 3,4-diMeOnitrosytrene to nitroalkane.
Haven't tried this anymore on the 3,4,5-trimethoxynitrostyrene though.
But what I noticed in my 3 experiments that I did was the following:
(shit haven't got my notes at hand now)
What I remember is that the method pointed out by vaguuh worked the best 97,5% yield.
Then I tried barium's method that was IIRC 93% and I used dioxane following a literature paper and got 78% or so.
I'll post the exact experimental data soon, what I do remember is that I added the nitrotyrene as a powder in the first two cases. The NaBH4 solution
was at 30*C constantly for 1,5 hours (took 45minutes to add the nitrotyrene).
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by Klute
Nice! Doesn't the PTC cause any problems with the nitro reduction? |
the PTC is removed by distillation. its bp. is 225°C@760mmHg.
i am not sure how it would poison a catalyst, though.
DNA: which method from Barium do you mean you got 93% with, on what substrate?
[Edited on 31-3-2008 by stoichiometric_steve]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
| Quote: |
the PTC is removed by distillation. its bp. is 225°C@760mmHg. |
duh... of course how stupid of me. I'm a bit elsewhere lately
At what vacuum did you distill your nitroalkane, i get it the mentionned bp is that of the PTC (which did you use BTW: TBAB, CTAB?
How does the solvant volumes compare to the EtOAc/EtOH? (though i think i remember you said you never tried that system before.. not sure) Abs EtOH
is time consuming..
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by Klute
At what vacuum did you distill your nitroalkane, i get it the mentionned bp is that of the PTC (which did you use BTW: TBAB, CTAB?
|
i used Trioctyl methyl ammonium chloride a.k.a. Aliquat 336, see above. This is about the cheapest PTC i think.
The nitroalkane i distilled came over at around 84°C@0.4mmHg.
the solvent volume, well, as stated:
you need approx 450ml toluene for 1mol, and the water phase was approx. 1L. less water could probably be used.
|
|
|
DNA
Hazard to Others
Posts: 191
Registered: 11-6-2003
Location: @moon
Member Is Offline
Mood: Experimenting
|
|
I used it on 3,4-dimethoxynitrostyrene and this is the one I tried
http://designer-drug.com/pte/12.162.180.114/dcd/chemistry/ni...
And then also vaguuh's method and a paper I got lying around.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
CTH according to Pasad et al.
According to Pasad et al, article kindly posted by StoechiometricSteve in the first page of this thread, nitroarenes are quickly reduced in
CTH conditions using formic acid and catalytic triethylamine. This forms triethylammonium formate in situ, which upon transfering hydrogene,
forms free triethylamine and CO2.
Altough they add the formic acid in 30min, they exclusively use nitroarenes, which are known to react much faster than aliphatic nitroalkanes. Thus,
when using nitroalkanes, it is advised to add the formic acid at a slower rate, to avoid acidic conditions.
StoechiometricSteve tried out a variation of this reaction, and told me that the reaction stopped if pH turned acidic.
I decided on trying this reaction out, but by using a larger amount of triethylamine, and adding the formic acid at a slow rate.
In the article, they claim that substrate or products that are basic enough to form formates can also act as a catalyst. When reducing nitroalkanes,
the formed amines are often basic enough to enter this catalysis cycle. An interesting twist would be using a catalytic amount of product at the
begging of the reaction!
2-MeO-nitroethane was used because it was in the fridge since a few months and i didn't have much use with it. It contained some dimer (obtained by
naBH4 reduction of the nitrostyrene) as shown by TLC.
Catalytic TRansfert Hydrogenation of 2-(2-methoxyphenyl)nitroethane with formic acid and catalytic triethylamine
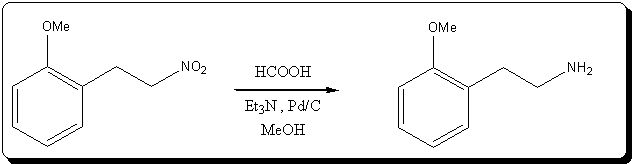
4.28g (23.91 mmol) of 2-(2-methoxyphenyl)nitroethane, a golden yellow viscous oil, was diluted with 10mL MeOH.
0.5g 10% Pd/C (Acros) were weighed and transfered to a 100mL 3-neck RBF flask, equipped with a condenser attached to a bubbler, a addition funnel, a
thermometer and a stir bar. It was wetted down with 70:30 MeOH: H2O under a flow of argon. The substrate was added with 20mL more MeOH, under slow
stirring. This formed a black suspension.



1,0 mL (7.21 mmol) of triethylamine (Acros) were added via syringe, and the flask was immersed in a 50°C oil bath.

4.6 mL (101.49 mmol) of 85% formic acid were then transfered to the addition funnel, and very slowly dripped in (1drop/40-50 sec). A slow, but
constant gas evolution started. There was no bubbling visible in the flask. The addition lasted two hours. No change in apperance of the reaction
medium. Heating and stirring were maintained for another two hours.
At the end of the addition, TLC (80:20:1 AcOEt:MeOH:Et3N) showed a large spot under the substrate, and only a small spot for the substrate. This
could very well be the dimer, as it is not seperated from the nitroalkane with this eluant system.
After the two hours stirring, TLC had not changed.
[Rest when i get time to upload pictures]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Unfortunaly, because of a stupid mistake, the other photos were lost... In any case, the reaction medium didn't change apperance, and the workup was
standard.
After 2 more hours stirring at 50°C, the flask was cooled down, and the black suspension filtered through two paper filter twice, giving a totally
clear filtrate [Note 1]. The cake was rinced with 2x20mL MeOH, and 10mL 50:50 MeOH:H2O [Note 2]. The now slightly milky solution was acidified with
conc HCl, causing the milkyness to increase. A little amount of clear, slightly yellow oil crashed out at the bottom.
The MEOH was removed at atmospheric pressure using a small coluum, until vapor temp increased over 90°C. The medium quickly took a blue (!) color
upon heating, and most of the clear oil turned to a black/blue viscous residu [Note 3].
The solution was transfered to a sepperating funnel, and washed with 3x25mL toluene, which removed the blue color, but left a milky slightly amber
aqueous layer. 20mL DCM wash removed a little of the color.
The aq was then basified with 15% NaOH slowly, causing strong milkyness and a clear oil to crash out. It was then extracted with 4x25mL toluene.
The combined opaque organics were thoroughly washed with 3x100mL 1% NaOH, which cleared it up pretty well, and 150mL brine, which gave a totally
limpid, very slightly amber solution. This was neutralized with conc. HCl (>2.5mL needed) and transfered to a 250mL wide-neck distn flask. A little
IPA was added to rince the erlenmeyer.
The IPA/Toluene/H2O azeotrope was removed by simple distn at atm, once all the IPA was removed, the distn setup was replaced by a Dean Stark, and
reflux maintained until no more water was collected and the head temp exceeded 100°C [Note 4].
Upon cooling, the slightly orange solution was covered with a little pet ether, which caused alot of milkyness, and a whitish solid appeared.
After cooling to RT, the suspension was filtered, and the white solid washed with acetone to remove some orange gummy material, then with pet ether.
It was dried by suction for 10min, then under a lamp to remove the solvant smell, then in a CaCl2 desicator. The crunchy cristalline mass weighed
0.94g (5.06 mmol) 21.07 % yield of 2-methoxyphenethylamine hydrochloride. The white crystals had a broad mp 139-146°C (but using a
very artisanal method: oven and thermometer  ), so it was not triethylammonium
chloride (mp= ~240°C) [Note 5] ), so it was not triethylammonium
chloride (mp= ~240°C) [Note 5]
The catalyst was regenerated by thoroughly washing with fresh 10% NaOH, then with water, sucking air through it for 10min, and washing it with a
little MeOH before leaving it dry overnight. It was easily scrapped off (though the filter paper obviously traps a certain amount). It is adviseable
to use the weakest vacuum possible when filtering it to avoid excessive plugging.
Notes
Note 1: I have to agree with Steve now, there is no need to acidify before filtering when using (tetralkyl)ammonium formates. This was a much easier
workup, and it avoids contaminating the catalyst with impurities comming from the acids (that's why i always used GAA and not technical HCl). But when
there's bicarbonates around, i guess it's better to acidify to remove them from the catalyst.
Note 2: This was to avoid a eventual fire hazard: it's is a known fact that 10% Pd/C can spontaneously ignite when wet with MeOH after a reduction.
The small amount of water prevents this, so the catalyst can be left on the buchner before being regenerated.
Note 3: I should have seperated what seemed to be unreacted nitroalkane before removing the MeOH... Heating for a certain period in acidic conditions
must have caused all kind of side reactions... I guess adding dilute brine and a quick extraction before removing most of the MeOH would give less
impurities..
Note 4: I'm not sure this is the best way for amine with ether groups: heating for a few hours (it is a long process) with excess acid at >100°C
temp could cause some demethylation? But it is so practical.... Using vacuum or pet ether could help keeping the temp down. I should just gas some IPA
with HCl i guess...
Note 5: The repeated water washes should remove small quantities of triethylamine quite easily (solubility : 7.5g/100g), but if using bigger amounts,
it should be seperated from the amine freebase by vacuum distn, or at least leaving the freebase under high vacuum long enough to remove most of it
without distn of the freebase itself.
Comments:
Well, the method surely isn't optimized for nitroalkanes yet. B ut it can be! I'm sure using at least 1 eq of triethylamine, or directly dripping 1
eq of triethylammonium formate, then 2-4eq of formic acid could be better, avoiding any acidic conditions, and ableing a quicker addition. The fact
that there seemed to be no excessive gas evolution could mean no wasted H2.
Seems promising to me. Ok, you could just aswell tell me go buy some ammonium formate or stick to the potassium formate, at least you would get
proper yields, but where's the fun in that? And in any case ammonium formate is
too expensive for me to not consider anything else
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
you should consider generating H2 from metal + acid. much cheaper than NH4COOH and most likely better yields. i will try it soon, and i'm getting
myself familiar with low pressure hydrogenation, too. i'll build a reaction vessel from a 2L duran bottle (Schott brand, those with the blue GL45
caps). i guess those hold well up to at least 3 bar.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Yes, that is a good idea. Do you intend on modifying yourself the lid, or do you have one with the gas inlet to handle dry reageants under inert
atmosphere?
Somehow, if i could find a easy, practical and good-yielding CTH procedure, i would be more confortable with it than a H2-pressurized vessel in
amateur settings, though.
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by Klute
Do you intend on modifying yourself the lid |
yes, but i think there are commercial tubing adaptors for the GL45 available.
|
|
|
beastmaster
Harmless
Posts: 38
Registered: 28-5-2008
Location: USA
Member Is Offline
Mood: hyper
|
|
RXN never took off
I have wanted to try out CTH, in place of Hydrogenation for several reactions. It seems so simple and safe, but each time I've tried it nothing has
happened. There is suppose to be gas generated from the NH4formate, as it decomposes into its elementary parts, NH4 H2 CO2.(or so I assume) Any way
could the problem be my Pd on Carbon? I bought it from a photo place. Should I or can I maybe pre-charge it with hydrogen before using it? I am
thinking this is where my problem lies, but I am at a lost. If I am not being clear it because of my lack of formal education. Any help will be
greatly appreciated. Thank you
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
If your Pd/C is already presaturated with H2, you will not observe any appreciable gas evolution when adding formate salts to it.
I have previously run CTHs with unreduced Pd/C, which generated lots of CO2 and heat, but those reactions also tended to produce quite an amount of
coloured side products (this is most likely since the catalyst dehydrogenates whatever it can grab from the reaction mix) and the catalyst was heavily
poisoned after a single run.
A recent CTH nitro reduction run (unknowingly) using presaturated Pd/C (9:1 MeOH:H2O, 5eq. NH4COOH, 20wt.% Pd/C 5%, stirred 24h) resulted in an almost
non-exothermic reaction, providing a quantitative yield of the amine.
Be aware that upon rinsing the hydrogen-saturated catalyst off the equipment, solvent vapours may catch fire. Be sure to flush the flask with whatever
inert gas you have or keep the catalyst wet with water.
You can presaturate your catalyst by simply letting it stir with some formic acid or formate salts, i.e. a mixture of HCOOH and catalytic amounts of
KOH (or NaOH or any organic amine). Bubbling will stop when the catalyst is saturated. It is probably a good idea to clean up the catalyst after each
reaction by repeated extraction with MeOH/EtOH/iPrOH to remove organic residues and keeping it in a dilute formic acid solution.
[Edited on 14-8-2008 by stoichiometric_steve]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
You could test your catalyst by adding a little formate solution to it, without any substrate, it should evolve some gas as Steve said. Better use a
bubbler to really see the gas evolution, rather than looking for bubbles.
When you say your reaction never took off, does it mean you recovered your starting material quantitatively, or simply that nothing seemed to happen?
As Steve explained, with saturated/activated catalysts, it seems only a true transfert hydrogeantion takes place, so no H2 evolution. Depending on
your reaction temp, the CO2 evolution might be unoticeable. It seems if the catalyst isn't saturated, it first form nacent H2 from the donors, absorbs
H2, and then reduces the substrate, so it's more of a dehydrogeantion/hydrogenation process.
Please giev use more details: what kind of substarte: alcene, nitro, etc, what solvent, temp, catalyst loading etc.
Be aware that most of the time, a CTH is just a black suspension, no excesive bubbling etc. It can look as nothign is happening, but that catalyst is
doing his job well
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Barium
Hazard to Self
Posts: 85
Registered: 24-8-2008
Member Is Offline
Mood: No Mood
|
|
Dear Steve, the aqueous solution of potassium formate forms one phase, the solution of the nitroalkane in IPA forms another phase and the catalyst
forms the third phase in the reaction mixture.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
What pleasure to see you here, Barium!
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
starman
Hazard to Others
Posts: 318
Registered: 5-7-2008
Location: Western Australia
Member Is Offline
Mood: No Mood
|
|
Welcome Barium! Been wanting to ask about RT ammonium formate/Zinc reduction of nitro compounds.Seems easier/cheaper and faster than anything
discussed in this thread.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Good luck with workup then
Apart from the price of Pd/C, this procedure is very easy to perform and employs cheap reagents. The workup is a breeze, and most of the time the
yields go from good to excellent..
I think Steve has some experience with Zn/formate reductions of nitro compounds, and should be able to comment much more than me on that subject.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
starman
Hazard to Others
Posts: 318
Registered: 5-7-2008
Location: Western Australia
Member Is Offline
Mood: No Mood
|
|
| Quote: |
Good luck with workup then
|
I take it that the work up is painful then?I saw Barium's write up of a Zn/formate reduction originally
published in the Indian journal of chemistry.Appears quite straight forward with short reaction times at room temperature.
|
|
|
| Pages:
1
2
3
4 |









