| Pages:
1
2
3 |
Alice
Hazard to Others

Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
@Assured Fish
| Quote: | I must admit you guys tend to loose me alot in this thread, i feel like the fumbling bafoon trodding along after you guys with pockets full of stones
 |
Just a matter of practice and experience. 
There are just so many things you don't think about if you don't have much experience (or a bad day ). That's normal.
Quote: Originally posted by CuReUS  |
isn't there a chance that some of the unreacted dihalopropane might also react with the succinimide ? |
Sure, but 1,3-diaminopropane is soluble in water while the product amine supposedly isn't. And it has reportedly a logP_ow of -1.3 (wiki, not sure if
this is calculated), which means it prefers the aqueous phase as the free base. Recrystallizaton of the substituted succinimides may be another way
separating it.
Here is a paper introduced showing a modified Grignard adding 3 mol% anhydrous CuCl2 in THF. Examples are:
p-methoxyphenylmagnesium bromide (1.2 eq.) with octyl bromide (1 eq.): 97% yield
p-dimethylaminophenylmagnesium bromide (1.2 eq.) with 1-chloro-5-bromopentane (1 eq.): 93% yield
With a bit of luck this works good as well with diethyl ether and the two possible reagents 1,3-diiodopropane and 1-chloro-3-iodopropane. CuCl2 is
about the most trivial additive I can think of and 3 mol% looks great. 
[Edited on 15-5-2017 by Alice]
EDIT:
confused edit removed
[Edited on 15-5-2017 by Alice]
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
p-chlorophenethyamine prep?
Ok so ive been searching around for a source of fenclonine and have found nadda, its like this stuff is a prescription med or something even though
all the sources ive read on the stuff don't refer to it being prescription based. 
So i propose a different route to synthesize pCPEA (p-chlorophenethyamine) based of the same concept but starting from phenylalinine.
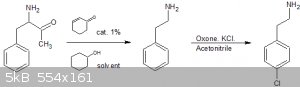
The decarboxylation is still the same as the one previously proposed by CuReUS here:
https://erowid.org/archive/rhodium/chemistry/trp.decarbox.en...
Then selective para-chlorination using potassium peroxymonosulfate (oxone) as a catalyst and KCl in acetonitrile.
https://erowid.org/archive/rhodium/chemistry/oxone.aromatic....
The only thing is I don't have direct access to the product Oxone, but i have found a product that contains peroxymonosulfate on ebay that looks like
oxone:
http://www.ebay.com/itm/saltscapes-chlorine-free-oxidizer-sh...
Also a source of phenylalinine that's pretty cheap: http://nootropicsdepot.com/dl-phenylalanine-powder/
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
@Assured Fish
Chlorinations (and halogenations in general) aren't compatible with amines, as it leads to chloramines. Just think about how the reaction works. Oxone
oxidizes chloride to chlorine, which is an electrophile and can be either attacked by the pi system of the arene or by the amine. But it may work by
protecting the amine. EDIT: Oxone may also directly oxidize the amine.
I was looking for a few possibilities avoiding CS2. The first couple of options involve the transformation of isonitriles into isothiocyanates by
different catalysts with elemental sulfur:
R-NC + cat. Se (5 mol%) + Et3N + S in THF, 73 % yield for nBuNC (Link)
R-NC + cat. Te (0.02 mol%) + Et3N + S in THF, 73 % yield for nBuNC (Link)
R-NC + cat. RhH(PPh3)4 + S in acetone (Link)
Selenium and especially tellurium are easy to aquire OTC as far as I see. Buying triphenylphosphine gives no fortune compared to buying CS2, making it
OTC is lots of work. The isonitriles can either be made from the amine and chloroform or from the halogen compound utilizing AgCN.
The next couple of reactions are thiocyanate <---> isothiocyanate isomerization based. Isomerizations lead to the thermodynamically more
favorable isothiocyanates - if they work:
thermal isomerization in solution (may not be suitable for non activated primary alkyl thiocyanates)
gas phase isomerization (reactive distillation)
heating alkylthiocyanate with metal thiocyanates
utilizing a lewis acid like ZnCl2
https://books.google.com/books?id=cAm7rA_D25wC
https://books.google.com/books?id=-OWIAwAAQBAJ
Third possibility:
reaction of alkyl halogenides with Hg(SCN)2 (possibly bad yield for primary alkyl halogenides, works at least bad for BuBr) (Link)
EDIT2: Would also be interesting how AgSCN works on primary alkyl halogenides, it wasn't examined in the paper above.
This is just what I found via superficial literature search. There may be more options of course.
[Edited on 16-5-2017 by Alice]
[Edited on 16-5-2017 by Alice]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Assured Fish | | Ok so ive been searching around for a source of fenclonine and have found nadda, its like this stuff is a prescription med or something even though
all the sources ive read on the stuff don't refer to it being prescription based. |
https://raypeatforum.com/community/threads/affordable-source...
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Two new drafts for the second part, the first for p-chlorophenylethyl halogenide the second for p-chlorophenylethylamine. I dont suggest any specific
chlorination for now, nor do I comment it, at least for now. 
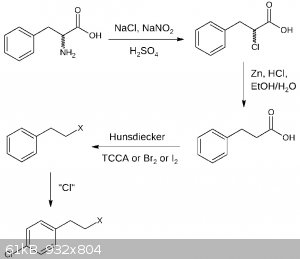
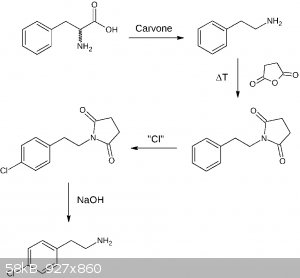
EDIT:
After I had a closer look at the first suggestion, it's a bit overcomplicated, so I made another draft:
The first step is taken from the literature, where NaBr is used (90% yield for phenylalanine). I speculate NaBr may be substituted by NaCl which hopefully works too. For the
chlorination the procedure posted by Assured Fish may work good here, a closer look at the original publication reveals for isobutylbenzene yields of 72% para, 2% ortho, and 1% disubstitution (75% conversion). Fairly good! The
substitution performed by AgCN is described in the literature, but I have to find a good source first. The decarboxylation hopefully works freely as
known for similar structures. The literature source for the last step is described in my previous post and can be found here.
[Edited on 16-5-2017 by Alice]
[Edited on 16-5-2017 by Alice]
[Edited on 16-5-2017 by Alice]
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
| Quote: |
Chlorinations (and halogenations in general) aren't compatible with amines, as it leads to chloramines. Just think about how the reaction works. Oxone
oxidizes chloride to chlorine, which is an electrophile and can be either attacked by the pi system of the arene or by the amine. But it may work by
protecting the amine. EDIT: Oxone may also directly oxidize the amine.
|
Fuck.
CuReUS i would incorage you to follow that link because its a dead end, even if you use the search engine on the site.
I did manage to find a source for fenclonine however its way over priced.
http://store.molbase.com/product-26118.html
Also please don't then give me this reference because I already checked it out and the product they are selling doesn't actually contain fenclonine,
they sell Centrophenoxine which doesn't contain any fenclonine, it contains another compound called Meclofenoxate which is an ester of
dimethylethanolamine and 4-chlorophenoxyacetic acid which is confusingly also named pCPA just to add confusion to a subject that the general public is
already ignorant about.
http://www.aip-health.com/pcpa.html
@Alice
Thanks for the correction, looks like a protecting group of succinimide/phthalamide is the best way to go about it then and it could still be
accomplished in 2 pots.
A small note though Alice regarding your first suggestion, the hunsdieker reaction requires the carboxylic acid to be converted to a silver, lithium
or thalium carboxylate first, I also am pretty sure it has trouble proceeding on saturated alkyl carboxylates, it would be better to decarboxylate and
then diazatize the amine followed by reaction with the halide salt to get the desired alkyl halide.
| Quote: |
I was looking for a few possibilities avoiding CS2. The first couple of options involve the transformation of isonitriles into isothiocyanates by
different catalysts with elemental sulfur:
|
Yusss someone else thinks the isonitrile is a better route than CS2, try telling CuReUS that; he thinks we're mad.
Edit: just realised that phthalamide would also be chlorinated so succinimide is really the best option.
[Edited on 17-5-2017 by Assured Fish]
[Edited on 17-5-2017 by Assured Fish]
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Assured Fish |
@Alice
Thanks for the correction, looks like a protecting group of succinimide/phthalamide is the best way to go about it then and it could still be
accomplished in 2 pots. |
Found a comment from Nicodem on a very similar problem. He mentioned protonation of the amine may be sufficient. This may have implications on the
type of chlorination reaction to be chosen. (Link).
| Quote: | | A small note though Alice regarding your first suggestion, the hunsdieker reaction requires the carboxylic acid to be converted to a silver, lithium
or thalium carboxylate first, I also am pretty sure it has trouble proceeding on saturated alkyl carboxylates, it would be better to decarboxylate and
then diazatize the amine followed by reaction with the halide salt to get the desired alkyl halide. |
It may work better for unsaturated acids but I don't think it's limited to such cases. Honestly I was just looking if it works for aliphatic acids but
didn't read into details. See The Degradation Of Carboxylic Acid Salts By Means Of Halogen - The Hunsdiecker Reaction:
| Quote: | | The greatest utility of the Hunsdiecker reaction lies in the preparation of aliphatic halides, and most of the investigations of the reaction have
been in the aliphatic series. |
Anyhow, I discarded the first route more or less, as like mentioned, it's a bit overcomplicated. About your second point, not sure if the reaction on
primary amines works as excellent as the one I found. I remember there are some issues with alkene and alcohol formation.
| Quote: | | Quote: |
I was looking for a few possibilities avoiding CS2. The first couple of options involve the transformation of isonitriles into isothiocyanates by
different catalysts with elemental sulfur:
|
Yusss someone else thinks the isonitrile is a better route than CS2, try telling CuReUS that; he thinks we're mad. |
We're all mad here...
| Quote: | | Edit: just realised that phthalamide would also be chlorinated so succinimide is really the best option.
|
Probably not, as it's deactivated. The authors of the paper you referred to (KCl-Oxone system) didn't find any chlorinated product for benzoic acid
for example.
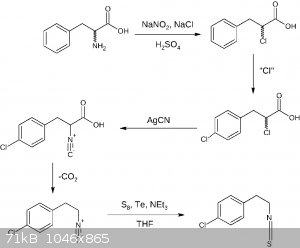
Here is the 4th draft assuming there is no need for amine protection other than protonation. The first two steps may be done the other way round,
additionally the second popular isocyanide formation reaction is shown, avoiding AgCN:
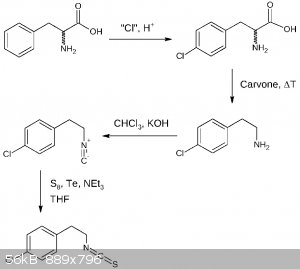
EDIT: As an additional note: THF utilized in the last step may be substituted by diethyl ether or dioxane making this reaction more OTC.
[Edited on 17-5-2017 by Alice]
[Edited on 17-5-2017 by Alice]
[Edited on 17-5-2017 by Alice]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
really ?
actually,the hunsdicker reaction gives excellent yields for saturated acyclic and alicyclic compounds.It gives shitty yields for aromatic COOH's
isonitriles and Te.What a wonderful combination to stimulate one's olfaction.
spring is going to come early for some this year
[Edited on 17-5-2017 by CuReUS]
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
I saw tellurium sold for element collections at ebay and as a money investment metal.
Good thing is as 0.0002 eq. are required, it makes 2.6 mg at 100 mmol scale.
[Edited on 17-5-2017 by Alice]
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
As I didn't mention this before, in my first route draft where I suggested cyclisizing the amine directly with the benzyl ether, displacing methoxide, it caused quite some doubts about how this
might be possible. This idea came to me as I remembered having read a paper about the synthesis of levetiracetam (An alternate synthesis of levetiracetam) in the past and thought about if there is a possible cut through for the present synthesis. For some
instances the case differs, but for others it doesn't, which I will explain in the following.
For the levetiracetam synthesis, 2-aminobutanol is reacted with gamma-butyrolactone whereas in the first step aminolysis with ring opening occurs
resulting in an amide. Then the amide-N acts as a nucleophile and substitutes the hydroxy group. The first step isn't really sursprising, while the
second one would probably not have been expected. It becomes more clear as in a former synthesis 4-chlorobutyryl chloride was utilized instead of
gamma-butyrolacton, a much more expensive and hazardous reagent. The reaction is performed at different temperatures, 160 °C (8 h, 76% yield, 5 mol%
H3PO4 as catalyst) and 225 °C (10 h, 93% yield, no catalyst) solvent-free. Additionally the reaction can also be enhanced by microwave irradiation
providing lower temperatures and greatly reduced reaction times (1 h, 200 °C, 82% yield). The reaction works because five-membered ring formation is
favored (Baldwin's rule), it even outperforms hydroxide being a bad leaving group and an amide being a bad nucleophile.
For the reaction I suggested, a less kinetically and less energetically favored 7-membered ring is proposed to form and the less favorable methoxide
as the leaving group. Nevertheless, two out of six angles are sterically fixed, making the 7 membered ring already partially in place, the benzylic
position is activated for substitution compared to an aliphatic position, and the amine is a good nucleophile. An interesting effect known for
benzylic substitution reactions is the dependence of rate constants on aromatic substitution pattern. While EDGs in para and ortho position enhance
reaction rates, destabilizing the bond between benzylic carbon and substituent and stabilization of the transition state. EWGs show the opposite
effect. (See: Kinetics and Mechanism of the Pyridinolysis of Benzyl Bromides in Dimethyl Sulfoxide or https://books.google.com/books?id=AgU4AAAAIAAJ&pg=PA134).
As I mentioned before my suggestion is, in my opinion, a very experimental suggestion, but didn't come from nowhere. The only question which matters for the proposed reaction is if it proceeds in a
considerable speed before pyrolysis occurs. H3PO4 or microwave probably wouldn't hurt too
[Edited on 21-5-2017 by Alice]
|
|
|
tsathoggua1
Hazard to Others
Posts: 335
Registered: 8-1-2017
Location: Beyond the pale
Member Is Offline
Mood: Phosphorescent
|
|
Having looked up fenclonine, I can more or less guarantee that, short of third-world or countries where pharmacists are bribable for more or less
anything if somebody has the money, given its action that isn't a drug that is going to be OTC. Nasty looking stuff too, very similar in structure and
action to the para-haloamphetamines, which, 4-fluoroamphetamine aside, are notorious serotonergic neurotoxins that cause profound, massive depletion
of tryptophan hydroxylase. IIRC at one time para-chloroamphetamine was originally developed, or certainly, used, as a diet drug, until it became known
quite what a noxious bunch of compounds they are. Certainly the alpha-methylated homologs are, and going from a quick glance at the wiki article,
fenclonine is also (deliberately) used to knock out serotonin production in cases of carcinoid, which is the term for a serotonin-producing tumor. Not
really the kind of thing one would pick up in a health food shop, nootropic vendor, or anything along those lines, or ingest without it being pretty
essential.
As for the production of 1,3-chloroiodopropane, I recall that iodine monochloride adds to double bonds to give chloroiodides. If iodine is available
or KI, NaI etc. then the synthesis would require only chlorine gas, I made a couple of oz of ICl a little while back actually and the synthesis is
quite easy, if a little hairy.
Its water reactive, air reactive, doesn't much like prolonged exposure to sunlight from what I've observed over time (it appears like the dregs in a
flask, left outside, since I haven't yet got round to unsticking some stuck joints on the still I used that have gotten gunked up with iodine and
possibly ICl in the solid phase (its got weird melting kinetics, reasonable nominal melting point, but it can seemingly supercool and remain liquid
for prolonged periods of time that are significantly below the supposed freezing point, plus two crystalline forms. I actually have mine stored in a
glass bottle with a chemical resistant cap and seal, not sure what plastic since I didn't buy them, the filth raided my place on a bogus warrant
ostensibly to 'take samples for analysis', and what they actually DID, was trash the place, place small quantities of various reagents in these
resistant bottles they brought, which they then left on the fucking bench top, along with some opened bottles of ether, THF and Li. Bloody moronic
cunts if they weren't deliberately trying to burn the house down (there is some history behind that thought, although I'll not go into too many
details)
So I don't know what plastic the caps and seals are made of, I'd guess some kind of fluoropolymer because ICl is damned aggressive towards many
plastics, although it hasn't attacked the teflon tape used to assist sealing the joints it made mincemeat of grease and of keck clips (they fell to
bits at the rate that I'd used all but four of mine within a day or twos work. Didn't do much good to the straps on one of my gas masks either.) The
dregs are still in the receiver flask, and it appears to dissociate in bright sunny weather to give greenish fumes within the still sealed flask, and
reform itself overnight or during not so bright weather. Definitely not something to inhale, I'd treat it like bromine, only quite a bit nastier, a
bit easier to confine and unlike bromine, it is very moisture sensitive, a drop into water dissociates instantly into sinking iodine particulate and a
greenish yellow puff of chlorine gas. Synthesis is trivial though, a chlorine generator connected to a drying tube packed with anhydrous CaCl2 with a
bit of sacrificial wire wool to hold the drying agent in place if using liquid-phase chemistry to generate your Cl2, and the dried Cl2 is run through
a condenser, horizontally, set up as a distillation rig. A little excess I2 is advisable, since any formed iodine trichloride (a yellowish to orangey
solid) that forms initially as a byproduct reacts with solid I2 in the condenser (I used two liebigs in series with one another, one to pack with I2,
and the second to condense the vapors and the recieving flask in an ice-salt-water bath with a little ethylene or diethylene glycol (Forget which now,
but this just serves to cool the recieving flask down so it isn't too important which, mainly because it really isn't something you want to work with
outside of a hood, unless you do it outdoors (which I did)
As I recall, it adds across double bonds like a halogen forming mixed chloroiodides. Whether or not the two halogens add proximally or distally to
each other on an allene I am not sure, but if the latter occurs to any extent then I would think propadiene, best diluted in a carrier gas such as
argon, bubbled through a solution of ICl due to the potent oxidizing nature of the beast, in GAA (which is a compatible solvent for ICl, Sasha Shulgin
(may his soul rest gloriously) used it during his synthesis of 2C-I for the iodination of 2C-H and he did so as a solution of the interhalogen in GAA.
would thus serve, IF distal addition occurs rather than forming an tetrahalopropane, or hexahalide
Then it might serve to produce the 1-chloro-3-iodopropane.
|
|
|
clearly_not_atara
International Hazard
Posts: 2788
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
I think you can add halogens to cyclopropane and get 1,3-halopropanes. In some literature the related 1,3-dichloropropEne is used as a 1,3-linker that
can attach each end separately.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
@tsathoggua1
propadiene exists at an equalibrium with propyne and therefore is likely to form 1,2 dihalogenated propane as a major side product. The other issue
with halogenating propadiene is how the fuck you would produce propadiene in the first place.
Atara's suggestion for using cyclopropane would be preferable given that it can be produced by reaction of sodium metal with 1,3 dibromopropane.
However this would again just push us back to having to prepare 1,3 dibromopropane. I reckon a better way to prepare 1-chloro-3-iodopropane would be
to first prepare allyl chloride by substitution of allyl alcohol with HCl in the presence of aluminium chloride, followed by distillation BP 45*C and
then carry our a reverse markovnicov using hydriodic acid followed again by distillation, the BP of 1-chloro-3-iodopropane according to sigma is 170 -
172*C, as for the BP of 1-chloro-2-iodopropane I am unable to find any references but 1-chloro-2-bromopropane has a BP of 116-117*C and
1-chlor-3-bromopropane has a BP of 144-145*C and this would suggest that the iodides would also have a sufficiently different BP so that fractional
distillation would be plausible.
https://en.wikipedia.org/wiki/Allyl_chloride
http://www.sigmaaldrich.com/catalog/product/aldrich/234478?l...
http://www.sigmaaldrich.com/catalog/product/aldrich/231274?l...
http://www.sigmaaldrich.com/catalog/product/aldrich/b62404?l...
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Seems like I don't get the point. What's the problem with chlorinating 1,3-propanediol by NaCl/H3PO4 followed by Finkelstein reaction?
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by tsathoggua1 | | Having looked up fenclonine, I can more or less guarantee that, short of third-world or countries where pharmacists are bribable for more or less
anything if somebody has the money, given its action that isn't a drug that is going to be OTC. |
why do you want to buy it from a brick and mortar shop,you could just order it online.Don't tell me you take the literal meaning of the word OTC
|
|
|
clearly_not_atara
International Hazard
Posts: 2788
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
https://www.google.com/patents/US3927131
I thought this was a thing.
Glycerol + 3 HCl >> 1,2,3-trichloropropane
"" + base + [rare earth metal oxide?] >> 1,3-dichloropropylene
| Quote: Originally posted by Alice | | Seems like I don't get the point. What's the problem with chlorinating 1,3-propanediol by NaCl/H3PO4 followed by Finkelstein reaction?
|
You get a mixture of dichloropropane, chloroiodopropane, and diiodopropane. If you don't mind losing at least half of your starting material along
with plenty of iodine it could be okay. Separating that mixture will require fractional distillation at least.
|
|
|
tsathoggua1
Hazard to Others
Posts: 335
Registered: 8-1-2017
Location: Beyond the pale
Member Is Offline
Mood: Phosphorescent
|
|
Curious-guilty as charged, in that instance. Thats having a literally-inclined mind for you, it does that sometimes and realizes later
That, and I am not sure how much I trust online pharmacies, particularly ones I haven't in the past done business with. There are certainly plenty of
skeezy thieving scumbags around, its just the sort of business that attracts such vermin like shit draws flies.
Also, whilst I don't know for sure, it looked like the kind of thing that might not survive the test of time, with regards to being in current use or
not, owing to the discontinuation of para-chloroamphetamine, which, unfortunately, was used as a diet drug, until it was realized that the
non-fluorinated para-halogenated amphetamines (and presumably para-cyano/4-nitro also) are seriously bad news and produce potential permanent or very
very long lasting depletion of tryptophan hydroxylase. Although at least, the phenethylamine homolog might prove to be a better substrate for MAO-a
without the alpha-substituent.
Didn't stop some bloody stupid and reckless online grey market vendors marketing para-bromo(meth)cathinone for some time, though. Which if its still
around, could be an alternative with reduction of the ketone and finkelstein reaction, if an alpha-methyl substituent (using the nomenclature for
numbering of phenethylamines and amphetamines) would produce an analog of capsazepine active at TRPV1 receptors.
As for the propadiene, its sold in my part of the world (UK of all the shitheaps) for use in welding. I hadn't realized about the equilibrium, or at
least, wasn't sure how fast they would interconvert and had thought to distill it under a freezing mixture (or rather, let it distill itself, without
external applied heat), the propyne in MAPP gas would come off first.. How fast do they interconvert? and what are the numbers like for the relative
proportions of propyne and propadiene at RT and in the liquid phase? Perhaps letting it boil off a solid, porous substrate such, ideally, as that from
an empty acetylene bottle since acetylene at least, doesn't particularly like to be stored in the liquid phase, although if not under pressure, would
that not largely mitigate any explosion risk? if in an open system that is.
(if nothing else, at least some of those absorbents use DMF as a solvent, some use acetone, and some DMF which wouldn't exactly be a crappy booby
prize for effort expended)
[Edited on 1-6-2017 by tsathoggua1]
[Edited on 1-6-2017 by tsathoggua1]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Actually most "MAPP" gas these days (at least here in the UK) is a propane/propylene/dimethyl ether mixture. Generally sold under the name "MAPP Pro"
or something similar, and ironically doesn't contain MAPP. I believe this substitute product was formulated following a shortage of MAPP a few years
back.
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
| Quote: | | You get a mixture of dichloropropane, chloroiodopropane, and diiodopropane. |
Sure, but boiling points lie sufficiently apart (120 °C, 170 °C, 224 °C). Dichloropropane can be removed without vacuum, 1-chloro-3-iodopropane by
aspirator vacuum, the diiodo compound remains and can be recycled or used for other reactions. I don't think this is a bad deal considering the easy
preparation and workup.
EDIT: erroneous logic removed.
[Edited on 1-6-2017 by Alice]
EDIT2:
I was indeed using a model not suitable here which gave me 75% yield.
After having removed some errors, here is my revised numerical result for 1 eq. dichloropropane and 0.9 eq NaI. In the first place I tried out 1 eq.
NaI, but it turned out this does no favor compared to 0.9 eq. and leads to a product ratio of 25% : 50% : 25% instead of 30% : 50% : 20% (rounded).
The overall iodide efficiency therefor is 56%. Trying 0.8 eq NaI I obtained the following ratio: 36% : 48% : 16% giving a NaI efficiency of 60%. A is
the dichloro, B NaI, C chloroiodo, and D is the diiodo compound. I hope this is free of silly errors now.
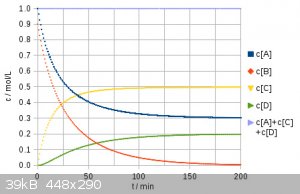
[Edited on 1-6-2017 by Alice]
[Edited on 2-6-2017 by Alice]
|
|
|
halogen
Hazard to Others
Posts: 372
Registered: 18-4-2004
Member Is Offline
Mood: No Mood
|
|
Azetidine might be an ideal way to introduce the C3 segment on to catechol, directly. Thereupon reaction with formaldehyde completes the cycle. More
precisely an acid salt would be used.
Who has azetidine lying around? This preparation in orgsyn uses ethyl acrylate to protect the amine. http://orgsyn.org/demo.aspx?prep=CV6P0075
I wonder if it is possible to prepare by anodic oxidation in dilute solution, of any gamma amino butyrate salt azetidine itself or aminopropanol.
Though oxetane is much less toxic/carcinogenic than oxirane, apparently not being an alkylating agent, the greater equilibrium of the amine salt in
the body could conceivably be relevant. I couldn't find anything to settle this, looking quickly. The relative difficulty of synthesis makes
azetidines uncommon - so says wikipedia.
[Edited on 9-6-2017 by halogen]
F. de Lalande and M. Prud'homme showed that a mixture of boric oxide and sodium chloride is decomposed in a stream of dry air or oxygen at a red heat
with the evolution of chlorine.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
@halogen How would you go about attaching the catechol ring with the azetidine, The amount of effort required to prepare the azetidine might make
other methods considerably more accessible.
|
|
|
halogen
Hazard to Others
Posts: 372
Registered: 18-4-2004
Member Is Offline
Mood: No Mood
|
|
That occurred to me. Actually, looking into it the O than the C seems more likely to be alkylated... in addition to the o/p problem. So, bad idea.
Sorry.
GABA electrolysis might just work, though, being necessarily dilute, separating it would be inconvenient. I should take out a patent. Come to think of
it, maybe the primary radical can be trapped by catechol, benzodioxole or something. That would be the final word in convenience.
[Edited on 9-6-2017 by halogen]
F. de Lalande and M. Prud'homme showed that a mixture of boric oxide and sodium chloride is decomposed in a stream of dry air or oxygen at a red heat
with the evolution of chlorine.
|
|
|
clearly_not_atara
International Hazard
Posts: 2788
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
On the subject of heterocycles, I thought of a way to use 2-aminothiazole in order to avoid using other sulfur compounds. Unfortunately, I'm not sure
how to synthesize 2-aminothiazole, although I don't think it's too hard. For the direct synthesis you can make a bromoacetaldehyde synthon via the
reaction of a vinyl ether with Br2 IIRC.
Anyway, step 1 is a Mannich reaction, step 2 is just the addition of p-chlorophenethyl iodide to the nitrogen.
Step 3 is ozonolysis: my guess was that alkylating the nitrogen makes thiazole lose its aromaticity, meaning the alkene can be oxidized. The resulting
dicarboxylic acid should eliminate to the thiourea.
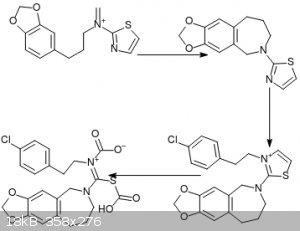
|
|
|
| Pages:
1
2
3 |
|








