| Pages:
1
2
3
4 |
CuReUS
National Hazard

Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by byko3y  | Cheddite Cheese, you would really wanted to read this doc before attempting to do clemmensen on the mannich base (which is a OC-C-C-N group).
Probably you've got an unsaturated compound.
|
I was expecting something to go wrong with the clemmenson,maybe a crazy polymerisation to yield a tarry goo or some ring contraction.But the
confidence with which cheddite extracted his product and purified it made me doubt myself,whether I was being too pessimistic
but mannich bases,decompose at temp above 120'C and I don't think the reflux temp went that high,did it ?
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Cheddite Cheese | | I finally got around to doing TLC. Compound before reduction is on the right, compound after reduction is on the left. Both compounds were spotted as
a solution of the hydrochloride. I used acetone to elute the plate, and alkaline permanganate as a stain. To get the stain to develop, I heated the
TLC plate on the hot plate at the lowest setting. The time between when the spots appeared and when all the permanganate had decomposed was only a few
minutes, but the spots were clearly visible as tan on a pink background, and I circled them lightly in pencil (sorry for the bad lighting in the
photo). |
When using amine salts for TLC analysis, you should also have ammonia or triethylamine present in your solvent system in order to liberate the free
base. The quick discolouration of the background sounds like you've not thoroughly dried your plate before staining (and the residual acetone is
reducing the permanganate) or your temperature is too high. I use a digital heat gun and typically with permanganate you're talking 30 seconds to 1
minute at 80-150 *C (depending on how easily oxidised the compounds are).
| Quote: Originally posted by Cheddite Cheese |
I wish I could have used a solvent less polar than acetone, but acetone was the least polar solvent I have that doesn't react easily with
permanganate. (I have toluene, but that would react.) Something like a mixture of hexanes and ethyl acetate (which I used in my organic chemistry lab
class) would be ideal. If these results don't meet the challenge expectations, I could try making some diethyl ether and using that.
|
Any solvent can be used for developing TLC plates, regardless of which stain you intend to use. The key thing to ensure is that the
plate is thoroughly dry before staining. This can be done by leaving it to evaporate for 5 mins, gently heating with a heat gun (ca 50 to 60
*C), or if your compounds may be heat labile, under reduced pressure (vacuum dessicator works well for this).
| Quote: Originally posted by Cheddite Cheese |
However, I think that these results show clearly enough that the two compounds are pure, that they are different, and that the compound after
reduction is less polar than the compound before reduction.
|
Not quite. Impurities can be pretty good at hiding, and I've seen such instances first hand multiple times (I'm a chemist by profession...). I have a
couple of guidelines that might help you out a bit though:
a) For a given solvent system, try to get your analyte close to the middle of the plate (by adjusting the ratios of the solvents). Ideally, we're
talking Rf = 0.3 to 0.7. The reason for this is that at low Rfs its difficult to clearly see separation, and at high Rfs several components can quite
easily "bunch together"
b) Visualise your plate by several methods. Unfortunately its not often possible to see everything using one approach. You should definitely have a
look under both short- (254 nm) and long- (365 nm) wave UV before using any stain.
c) Evaluate your compound using several different solvent mixtures. I've had cases recently where two compounds almost co-elute in several different
solvent systems (they fluoresced different colours under long-wave UV), but was fortunate enough to find a system that did provide separation. You
have several options depending on the polarity of your analytes; you'll get a feel for whats "about right" as you gain experience.
Please bear in mind that this advice is general and intended to be helpful.
| Quote: Originally posted by byko3y | Cheddite Cheese, you would really wanted to read this doc before attempting to do clemmensen on the mannich
base (which is a OC-C-C-N group). See attachment below.
Probably you've got an unsaturated compound. You didn't do the melting point check on the resulting amine, it would probably show if you've got the
right substance. Also there's a successful synthesis of 4-oxo-xxxpyperidine and it's subsequent oxidation on this forum http://www.sciencemadness.org/talk/viewthread.php?tid=3960&a...
|
I've glanced over the attached paper and found that all the examples are benzylic ketones; it may be that the mode of reaction is specific to that
structural motif. Whilst it is worth bearing in mind, I don't think we'll see any unsaturated species using triacetoneamine as substrate. I do however
strongly second your suggestion to run a melting point, especially if theres a literature value for triacetoneamine hydrochloride.
[Edited on 23-3-2015 by DJF90]
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
I feel so stupid now for not realizing that I could use a different solvent, and it would be fine because it would evaporate. I did, however, let all
the acetone evaporate before staining. The quick staining and decomposition is probably a result of the level of heat I used.
I'll do the TLC again with toluene and triethylamine. Permangante and iodine are the only stains I have right now. Iodine wouldn't react very well
with the compounds I think I have, but if an alkene was present, it would show it.
I don't have UV lamps. (I did have one about a year ago, but the ballast quit working.) I can do a melting point check, but I don't have a dedicated
melting point apparatus. I'll have to use an oil bath.
|
|
|
Etaoin Shrdlu
National Hazard
Posts: 724
Registered: 25-12-2013
Location: Wisconsin
Member Is Offline
Mood: Insufferable
|
|
Very much welcome, that was an excellent first post.
That paper is interesting, not something I'd heard about before. For the statement that Clemmensen reduction of Mannich bases tends to produce alkene
byproducts, they reference Reduction: Techniques and Applications in Organic Synthesis, by Robert L. Augustine, pp. 186-194. I'll ask in References as
well, but has anyone got a copy of this and can scan or summarize it? I would be interested to know if it discusses substrates where the ketone is not
benzylic. Supposedly there's one on Amazon for $20 but I'm a bit nervous of buying it when the others are all around $215.
[Edited on 3-23-2015 by Etaoin Shrdlu]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
I don't think that toluene itself will move the analytes much - use it as the non-polar component of a mixture (perhaps with EtOAc or acetone - try
1:1 first for an idea of which direction to move in). Also, go easy on the triethylamine - 0.1%v/v ought to do it. You'll have to heat the plate
(gently) after elution to get it to bugger off though.
| Quote: Originally posted by Cheddite Cheese |
Permangante and iodine are the only stains I have right now. Iodine wouldn't react very well with the compounds I think I have, but if an
alkene was present, it would show it.
|
Both are good general stains. Iodine will typically stain most things by dissolution in the organic substance (giving dark brown spots on a light
brown background), whereas reaction with the analyte will give white spots.
| Quote: Originally posted by Cheddite Cheese |
I don't have UV lamps. (I did have one about a year ago, but the ballast quit working.) I can do a melting point check, but I don't have a dedicated
melting point apparatus. I'll have to use an oil bath. |
They aren't too expensive and I've posted before on this...
| Quote: Originally posted by DJF90 | For those in the UK, you can buy a counterfeit money detector from Maplin (http://www.maplin.co.uk/p/professional-3-tube-money-detector...). This is an updated model to the one I purchased a couple of years ago, which
came with a blacklight bulb which is typical for long wave detection (365 nm ish...). This new model claims to have two UV bulbs and a white bulb
also, so perhaps it already has detection at 254nm also. Otherwise, I purchased a 4W germicidal bulb on ebay that will fitted my lamp, allowing
detection for plates using a 254nm fluorescent material.
|
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
TLC (round 2)
Eluent this time was 40% acetone, 59% toluene, 1% triethylamine. I used minimal heat when developing, so I managed to get a picture in which the stain
had not faded (because of this, there are a few drops of stain solution left on the plate, and although they obscure the bottom part of the plate in
the picture, there are no spots in that area.)

Rf for compound with suspected ketone group: 3.2/7.0 = 0.46
Rf for compound after reduction: 4.8/7.0 = 0.69
[Edited on 23-3-2015 by Cheddite Cheese]
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
Good catch, DFJ90, as expected from the consistent high quality of your posts. Cheddite, as noted, you had it right with the free bases in your
earlier post. Ammonia is good (gaseous or aqueous choice can theoretically change solvent characteristics), though I have seen hydroxides used before.
Drying is important with the triethylamine you used as well, since that can sometimes show up with stains. When not used in the actual solvent system,
I have seen people go nuts over this mysterious impurity (starting material didn't require it in that case). You can get doubling or streaking of
amine spots otherwise due to protonation equilibrium. In the same manner, a carboxylic acid may require addition of a stronger acid to shift towards
the protonated form.
I haven't read the paper yet, but I second/third a melting point. If it's broad and/or depressed, you should try a 2D TLC. They are very useful. Very
rare to just pick one solvent system for TLC and call it a day. And I actually prefer iodine for staining, often. One useful aspect is that it can be
very labile and volatile, and so with time, a second stain can be performed if your organics are stable. Sometimes doing a second stain, using a UV
lamp, or running a 2D TLC will show a surprise before you get a MP, which can be more of an issue when checking column fractions.
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Sigma-Aldrich (http://www.sigmaaldrich.com/catalog/product/sigma/t4694) lists the melting point of 2,2,6,6-tetramethylpiperidine hydrochloride as 299-301 C. I
have currently no way of measuring temperatures that high (the highest I can measure is 250 C), so a melting point determination is out of the
question. I might be able to prepare a different salt, and find a reference for that.
I also found Sigma's listing for the 4-oxo-2,2,6,6-tetramethylpiperidine hydrochloride (http://www.sigmaaldrich.com/catalog/product/fluka/87910), which cites a melting point of 198 C with decomposition. I'm not sure how
useful that is.
I really wish I could just take an NMR spectrum.
[Edited on 23-3-2015 by Cheddite Cheese]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
2,2,6,6-tetramethylpiperidine freebase boiling point is 152 C. Actually, in the first post I wanted to say "boiling point" instead of melting.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Cheddite Cheese | Eluent this time was 40% acetone, 59% toluene, 1% triethylamine. I used minimal heat when developing, so I managed to get a picture in which the stain
had not faded (because of this, there are a few drops of stain solution left on the plate, and although they obscure the bottom part of the plate in
the picture, there are no spots in that area.)
Rf for compound with suspected ketone group: 3.2/7.0 = 0.46
Rf for compound after reduction: 4.8/7.0 = 0.69
|
Much better, well done. It looks like you're sat on the floor to take that picture?
Cheers. I've considered writing a blog or a web page or something but just don't have the time.
| Quote: Originally posted by Chemosynthesis |
Very rare to just pick one solvent system for TLC and call it a day. And I actually prefer iodine for staining, often. One useful aspect is that it
can be very labile and volatile, and so with time, a second stain can be performed if your organics are stable. |
I used to use iodine as a preliminary stain but found that sometimes it can be difficult to fully remove. I do however find it is a good general
stain. I tend to shy away from permanganate as the LOD (limit of detection) tends to be quite poor. I prefer ceric ammonium molybdate or
vanillin/anisaldehyde as a day-to-day stain.
| Quote: Originally posted by DJF90 |
b) Visualise your plate by several methods. Unfortunately its not often possible to see everything using one approach. You should definitely have a
look under both short- (254 nm) and long- (365 nm) wave UV before using any stain.
|
I took the liberty of taking some photos this morning in the lab to illustrate this. Coincidentally, this is the same reaction that I was discussing
in point c) in the above (quoted) post, and 10%v/v MeOH-PhMe as eluent was the only system that gave visible separation between the starting material
and desired product (ArCl and P on the plate, respectively). Images are of the same plate visualised under 254 nm, 365 nm and with permanganate (left
to right)
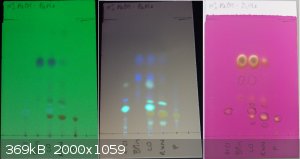
EDIT: The lanes marked ArCl, BPin and P contain more than one spot at 365 nm due to decomposition of the materials in solution (these are TLC samples
I've made up and clearly they don't store very well).
[Edited on 24-3-2015 by DJF90]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Is it a regular paper? Because mine glows only slightly blue under low pressure mercury ozoneless UV lamp (254 nm).
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Plans for oxidation to TEMPO
I'm adapting the procedure Klute used, given here: https://www.sciencemadness.org/whisper/viewthread.php?tid=39...
I've concentrated some hydrogen peroxide solution, and I estimate the concentration to be about 30%. I've measured the density as 1.13 to 1.14 g/mL.
Here's what I plan to do:
(I just found Klute's 2nd procedure, and I'm going to adapt that instead.)
[Edited on 29-3-2015 by Cheddite Cheese]
[Edited on 29-3-2015 by Cheddite Cheese]
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
That's a shame.
| Quote: | | I used to use iodine as a preliminary stain but found that sometimes it can be difficult to fully remove. I do however find it is a good general
stain. I tend to shy away from permanganate as the LOD (limit of detection) tends to be quite poor. I prefer ceric ammonium molybdate or
vanillin/anisaldehyde as a day-to-day stain. |
Thank you for the recommendations. I have been interested in ceric ammonium molybdate. I haven't ever used it before.
| Quote: Originally posted by byko3y | | Is it a regular paper? Because mine glows only slightly blue under low pressure mercury ozoneless UV lamp (254 nm). |
Nope. There is often an added fluorescent dye to the plates. I hate the un-backed paper for most purposes.
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Ok, so I finally have time to do this (I've been swamped with other work). If everything works, I'll be done before the deadline.
I'm planning to use sodium tungstate as a catalyst, adapting the procedure shown in the Science of Synthesis book.
Here's what I plan to do:
Dissolve
1 g (5.6 mmol) of 2,2,6,6-tetramethylpiperidine hydrochloride
0.25 g (6.25 mmol) of sodium hydroxide (for deprotonation)
0.47 g (1.42 mmol) sodium tungstate dihydrate
in 10 mL methanol and 4 ml water
Add
3.4 mL (33.5 mmol) of 30% hydrogen peroxide
Stir for a day, then extract with chloroform (I don't have ether), and evaporate the solvent under vacuum.
Edit:
The reaction has begun! Upon addition of the peroxide, the solution started to turn yellow. (Pictures will come this afternoon.)
~4 hours after reaction start: a yellow-orange color
[Edited on 22-4-2015 by Cheddite Cheese]

|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Workup results
0.535 g orange-red viscous oil (I was hoping for a nice solid  ). I might have to
do what the authors of the procedure did and use chromatography. ). I might have to
do what the authors of the procedure did and use chromatography.
Extraction was with 3x 5 ml portions of chloroform (the picture is of the first extraction). The portions were then combined and the chloroform
evaporated under vacuum.
The color of the oil is a bit redder than it appears in the picture, although it may depend on the lighting.
 
[Edited on 26-4-2015 by Cheddite Cheese]
|
|
|
Etaoin Shrdlu
National Hazard
Posts: 724
Registered: 25-12-2013
Location: Wisconsin
Member Is Offline
Mood: Insufferable
|
|
Awesome! Too bad it didn't crystallize for you.
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Column chromatography results: success!
I purified the liquid by column chromatography (200 mesh silica gel, ethyl acetate/toluene 50:50 eluent) to get 0.354 g of solid product. It's not in
the form of nice crystals, but I think it's reasonably pure. I measured the melting point as 31-37 C. With TEMPO's low melting point, I'm not at all
surprised that it wasn't solid until I purified it.
I think the 200 mesh silica gel that I used was a bit too fine, making the flow rate too slow and thus the band broader, but it was all I had. 200
mesh is best for flash chromatography.
 
[Edited on 29-4-2015 by Cheddite Cheese]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Please tell us the procedure you used for purification. Did you use a warm eluent? Did you use a paper chromatography results to understand the order
of eluates?
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
The eluent was room temperature. I didn't do paper chromatography on the crude product; since I knew the product had a red-orange color, I just
collected the red-orange band. I didn't see any other colored bands. The starting material, which I suspect to have been the main impurity, is
colorless.
My focus in doing the chromatography was not to analyze the crude product but to purify it, and I think it performed reasonably well.
|
|
|
Etaoin Shrdlu
National Hazard
Posts: 724
Registered: 25-12-2013
Location: Wisconsin
Member Is Offline
Mood: Insufferable
|
|
I missed the procedure to get OTC sodium tungstate. Regardless of that minor quibble, you've pretty clearly won here. The melting point is close to
what I'd expect for slightly impure TEMPO, and I don't think anyone else is going to squeak by on a technicality within the next few hours.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
I made TEMPO just now !
Simples : just go 1,2,3,4 and start drumming your fingers an sing or hum a bit.
Just kidding.
Congratulations to the Cheddite !
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
I think given the time limit (tomorrow), your characterization is relatively sufficient. Another TLC, possibly a 2D, or a reaction with a carefully
selected substrate would be pretty definitive for me on the basis of a home/hobby chemistry setting.
Considered using your presumed TEMPO for anything yet?
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Sodium tungstate can be made from tungsten, which, given your example of silver being acceptable, I think is fine. Since only a small amount of
tungstate is needed, someone could probably make it from light bulb filaments.
I can make sodium tungstate, if you need confirmation.
|
|
|
Etaoin Shrdlu
National Hazard
Posts: 724
Registered: 25-12-2013
Location: Wisconsin
Member Is Offline
Mood: Insufferable
|
|
No, you don't have to make it. Send me your Paypal or other prefered method of prize acceptance. 
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Well done, CC.
|
|
|
| Pages:
1
2
3
4 |








