| Pages:
1
2
3
4
5
..
26 |
CycloKnight
Hazard to Others

Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Electric benzaldehyde
Yes, i agree that there is a vast excess of oxidant. Though, it is perfectly regenerable and not too much of a bother - but scaling up is a
problem...for single batch runs. My cell holds exactly 2.2 litres of acid-oxidant mix, I don't think it's alot of work for 100ml - though
that does depend on what you want it for. It's really just a case of switch on, and let stir on the stirrer plate. A couple days later react and
extract. Practically a one pot synth, and able to produce all kinds of compounds. If you need such a unit to produce formaldehyde, why not use it to
make benzaldehyde as well? Or benzoic acid? I did a little bit of critical path management, and I ended up with the above method as the path of least
resistance (For me, at least).
Also, should you get busted - your lawyer could always argue that you use the cell for making formaldehyde...Or whatever, there is a long list to
choose from.
I am thinking of working on some kind of 2 phase oxidizing mixture, whereby the cell (stirred) is continuously run with a very large excess of toluene
floating on top which is continously removed and extracted (distilled).
Excess toluene simply being returned to the cell. The benefit of this is that the cell runs constantly, but only needs a relatively TINY amount of
acid-oxidant mix (tiny compared to industrial sized single batches). Intead of having the large excess of oxidant that sits in the bottom of the
beaker, only enough that dissolves is necessary here, since it's continuously regenerated!
Larger current means more mix (or larger + electrode) - but you see my point. Also, the lead electrodes do not corrode. At least not to an obvious
extent - they change from shiny to grey.
Over time alot of benzaldehyde could be made and the process could be made mostly maintenance-free. Occasionally acid and oxidant carryover will need
to be removed from the benzaldehyde extractor/distiller, but that's not much maintentenance. This process (if workable) is mainly limited by the
anode surface area and the amount of current you are able to supply! (and corresponding cooling of course...)
However, after a trial run it may be found that the oxidant needs to be toluene-free when it is regenerated by the anode, which would mean a separate
vessel (with heating and cooling inbetween to drive off toluene prior to regneration) for regenerating. I suspect that benzoic acid by-product could
be generated if toluene is present in the mix while current is passed - but I don't know yet.
Maybe someone else knows?
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
Pictures!
Yes, let's see some pictures. I always enjoy gazing upon others' DIY setups. Upload to the "scipics" account <A
HREF="http://www.sciencemadness.org/talk/viewthread.php?tid=603">as described here</A> so that you can place multiple, inline
images in your post instead of using attachments.
PGP Key and corresponding e-mail address
|
|
|
Tetramer
Harmless
Posts: 2
Registered: 25-1-2005
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Icarus
There is another oxidant worth trying: persulphates are used in electronics etching and easy available. I read a patent that used persulphates and
compounds of copper, silver, or iron as catalysts for the toluene->benzaldehyde. I tried without succes before using
ammonium persulphate like patent.
Tetramer, I am not quite sure of your answer.
Are you stating that this reaction was successful? |
Reaction worked some. I smell benzaldehyde. I was unable to isolate it but I think I make mistakes. Persulphate is oxidant and other metals are
catalyst. I use ammonium persulphate since I had not sodium persulphate. Maybe this is part of my trouble. I want to try again but cannot find lost
patent 
I wasn\'t born a threat, but I was born to be one.
|
|
|
Organikum
resurrected
Posts: 2337
Registered: 12-10-2002
Location: Europe
Member Is Offline
Mood: frustrated
|
|
Posted by Osmium at the-hive:
| Quote: |
Benzaldehyde
Ferrous-Copper Catalyst: Toluene (7.6 g.), water (35 ml.), ferrous sulphate
(0.110 g.) heptahydrate, cupric acetate (0.072 g.) and methanol (8 ml.) are
placed in a 250 ml. reactor.
Sodium persulphate (47.05 g.) in an aqueous-methanol solution of sodium
persulphate is added slowly to the mixture which is maintained at 70.degree.
C., in an atmosphere of nitrogen and under agitation.
The organic phase is separated after two hours and the aqueous phase is
extracted with ethyl ether.
The combined organic phases are distilled to afford 8.29 g. (95% yield) of
very pure benzaldehyde (compared against a pure sample).
US-Pat 4,146,582
|
/ORG
PS: Ammonium-persulfate should work too. Pleasse notive the big amount of persulfate needed. Smell of bitter almonds doesnt say much - it smells
already in strong in traces in special when warm and with water.
[Edited on 27-1-2005 by Organikum]
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
misc.
NiO2: Eng. Pat. 22887, 1900
MnO2, H2SO4, and catalyst: GB138999
Belongs in the chlorination thread, if anywhere: GB816253
H2O2 and catalyst: US3531519
benzyl bromide and sodium nitrate: US1272522
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
{img}http://www.sciencemadness.org/scipics/anode.jpg{/img}
[Edited on 29-1-2005 by CycloKnight]

|
|
|
Icarus
Harmless
Posts: 12
Registered: 17-12-2004
Location: The Stratosphere
Member Is Offline
Mood: getting warmer
|
|
"PS: Ammonium-persulfate should work too. Pleasse notive the big amount of persulfate needed. Smell of bitter almonds doesnt say much - it smells
already in strong in traces in special when warm and with water. "
Someone who has forgoten more about chemistry, than I know about it, once said that there may be issues with the Cu salt and NH 3 complexing.
Only one way to find out I suppose.
[Edited on 28-1-2005 by Icarus]
[Edited on 28-1-2005 by Icarus]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Manganese ammonium alum cell
Okay, as promised here is my photo essay of the mananese-ammonium alum cell, which can be used for the oxidation of various compounds.
Here is the anode:
(before dark red oxidizer is electrolytically generated, this picture was taken after reaction was complete)

Here is the power supply:

Here is the overall setup:

The white powder in the coffee jar is the ammonium sulphate I made from the brown garden stuff using the simple purification procedure I described 2
posts ago in this thread.
Here is the cell a few hours after the reaction has commenced:

It took about 3 days to convert all of the yellow mangano-ammonium sulphate into the dark red manganese ammonium alum.
Reaction takes place with vigorous stirring (50 degrees C), the 2 phases MUST be mixed for reaction to occur, I used magnetic and manual stirring to
whip up the mixture. I let the reaction proceed for 1 day before finishing it up by heating to over 60 C and applying vigorous manual stirring with a
nickel lab spatula.
(note the organic top layer, due to incomplete mixing)
During reaction (sealed with foil to minimise losses):

The orange tinge to the toluene is indicative of the reaction progress. Not sure what the tinge is, condensation products of some kind I suppose?
Reaction is now complete:

Extraction of the goods:

Extraction almost finished!! :
[img]http://www.sciencemadness.org/scipics/last blob.jpg[/img]
Total organics extracted (including a few hundred ml toluene used for extracting):

Vacuum distillation:

Product:

Cell is ready for re-use:
Eggnog anyone? Just kidding - don't drink that!

It is important to get as much of the organic material out of the cell as possible, otherwise it will get oxidized to the acid. This cell smells a bit
like formic acid, due to benzoic acid being gererated when the current is switched on. It may be wise to do the final extraction with something like
ether, so it will boil off easily after wards. Not so with toluene, enough always remains to interfere with the regeneration process for a while.
Let us have some more!
Sir yes sir, regenerating right away sir!

Current of 2 amps (at 5 volts) is flowing through the mix.
The black floating bits have nothing to do with this process. They are bits of carbon (dehydrated organics), the result of another experiment that I
carried out immediately after the toluene oxidation run. Please ignore them.
[Edited on 30-1-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Yield
Okay, I would just like to say that the yield was a little on the low side, however please take the following into account:
1) Toluene wasn't toluene, it was a mixture of toluene, hexane and water.
2) The product was not washed prior to distillation, so I ended up with quite alot of black polymerised gunk (which appeared out of nowhere) in my
flask.
In fact, I ended up with more gunk than product, oops. I thought that by using a vacuum this time I could avoid the high temperatures which would
cause the product to degrade - I was mistaken.
The toluene I used for the extractions was wet and had a boiling pt of around 86 C. Since the cell is mostly sulfuric acid it dried it (which I
thought was a bonus!), so I didn't want to wash it - I wanted to recover dry toluene for other purposes.
See what happens when one takes shortcuts? So, most of my product was ruined, oh well - I'll just have to wait another 3 days...
I should also say that toluene is a bit hard for me to get. I've been using this OTC solvent formulation called "Evo-Stik Cleaner 191".
Evo-Stik is a contact adhesive used in the UK, but the cleaning formulation is a mixture of solvents (and water too!). The solvents are toluene,
hexane and small amounts of other solvents.
For the reaction carried out in the photo essay above, I used 250ml of Evo-Stik solvent. Then I used a further 150ml or so for the extractions -
though I could have extracted a bit more!
I did the extractions by adding the "toluene" to the 3L beaker, mixing it up real good and then just removing the top layer with a dropper.
Doing it that way removes the need to pour the whole mix into a separating funnel and shake. An unnecessary risk (in my opinion).
I would say that as long as everything has been done correctly, the yield is much higher than what I managed, though I can't give a figure
because of the lack of control conditions. My last cell contained an unknown quantity of oxidizer, and this is the first run with my new cell! So,
this is the first run I've carried out in about 2 years.
I can say that practically no benzoic acid is produced is the temperature if kept at 50 C and no higher.
The benzaldedhyde odour begins from about 30 sec after the toluene is added to the oxidizer mix.
The reaction takes several hours to complete. The heater on my hotplate isn't functional right now due to a damaged thermocouple probe, so I had
to keep putting it on the stove ever few hours to heat it up a bit. My other hotplate has a weak stirrer and can't properly stir the mix...
In a couple days or so, the cell should be regenerated and I will try again and post the results here.
[Edited on 30-1-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Okay, I did another run last night and it was a success. However, it was only a partial run.
I estimated that the cell was no more than 25% regenerated and I managed to get 25ml of benzaldehyde from the mixture. The cell was only medium pink,
as opposed to dark dark red when it is fully regenerated. The partial regeneration is due to organic acids which have been generated when the mixture
is heated over 60 C.
I washed the toluene extracts twice, and there was negligible degradation of the benzaldehyde during distillation, and no scent of benzoic acid.
I started the distillation under normal pressure, as soon as the toluene was mostly gone (I let it run to 115 C) I then cranked up the vacuum and
boiled off the remaining benzaldehyde until orange distillate just started to distill over.
There is a small problem I'm trying to work a solution for. Steam distillation is the way around it, but I want to avoid it at all costs.
The problem is that as the cell regenerates, some of the dark red oxidizer forms clumps on the anode, these clumps break off and settle to the bottom.
During the reaction these clumps of oxidizer don't react (unless they have first been pulverized) since they aren't dissolved.
After the toluene has been reacted and the reaction is finished, I have been heating up the mixture to drive off the excess toluene before
regenerating. What has been happening is that the "clumps" have then been reacting at the higher temperature with residual toluene to form
benzoic acid which stays in the cell.
I used to use steam distillation, which would just steam distill out all of the garbage, but it also leached out the cell acid - so I am keen to avoid
it this time.
The benzoic acid interferes with the regeneration process by acting as a competing current carrier; some of the current produces dark red salt, and
some of the current produces oxygen due to the benzoic acid. Obviously killing the cell efficiency and lengthening the regeneration time considerably.
This evening, I plan to try an ether extraction of the mix, and failing that - I will dump the whole mix into my 3L RBF and steam distill it.
I've never tried ether before, I think it is worth a try.
Steam distilling really does drive out all the oils and junk which can accumulate in the cell after a while, but it is not convenient for everyone.
One solution would be to use a glass rod with a flat end to pulverize all oxidizer before reacting with the toluene.
That way, no benzoic acid will be generated when the mixture is heated to drive off any excess solvent.
Another may be to use a more inert solvent (i.e. diethyl ether)to remove the residual toluene before heating it up. That way, the ether should just
boil off without reacting with any excess oxidizer still in the mixture.
Or perhaps, the normal technique I've been using could be applied, the benzoic acid is allowed to form as usual during heating, but is extracted
out with another (inert) solvent just before regeneration.
I dare say that another possibility would be to apply a vacuum to the mixture to drive out excess toluene, intead of heating it!
As soon as I have found a working way around this (and it will be soon), I will post more pictures.
|
|
|
frogfot
Hazard to Others
Posts: 212
Registered: 30-11-2002
Location: Sweden
Member Is Offline
Mood: happy
|
|
That was very interesting, CycloKnight. One thing I wonder, can you tell the amounts of MnSO4, (NH4)2SO4 and 60% H2SO4 that you've used to
prepare manganese ammonium alum? It's quite ignorant question but I'm low on the precursors and wanna do this with bigger yields.. Also the
patent didn't describe this in detail eather..
If I understand correctly manganese ammonium alum is MnSO4*1/2(NH4)2SO4, right? Couldn't one than simply use a mixture of these two salts? They
dissociate in water anyway..
Extracting with a solvent would be very convenient nearly for anyone.. Instead of ether one could use ethyl acetate of low boiling petroleum
distillate..
Only thing I could suggest to the synth is using a mechanical stirrer overhead instead of magnetic..
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
For the manganese-ammonium sulphate mixture, the ingredients I have used are based on the patent ratios and are approximately as follows:
680g MnSO4
310g (NH4)2SO4
700g H2O
1850g 91% H2SO4
this requires approximately 120 amp hours to fully regenerate.
These are the figures I recorded, though I had carried out an "on the spot" conversion of the patents 98% acid ratio to my 91% ratio, since
91% was all the hardware store had.
But a little bit more ammonium sulphate was added, and I've since had to add a little bit more water(200 ml approx). If the acid concentration is
too high, you will know because only hydrogen and oxygen will form at the electrodes, no dark red oxidizer at the anode. Even if you have alot of
organic acid in the mix, you will still see SOME red oxidizer forming so if you don't see any - then add more water.
I'm unsure why I had to add more water to get the cell to produce oxidizer, it is either because more sulphuric acid was generated in the cell
(perhaps from excess sulphate), or the organic acid that was produced after the first run adds to the overall acidity in such a way as to require
dilution in order to produce oxidizer?
I made the mixture up by first adding the ammonium sulphate to the acid/water(the acid was added to only about half of the water) mix, allowing it
all to dissolve. Then to that I added the manganese sulphate, the mix will then turn yellow as the manganese-ammonium sulphate precipitate forms. Then
I added the rest of the water. One piece of advice here - grind up the manganese sulphate BEFORE adding it to the ammonium sulphate/acid mix. Or you
will spend all night trying to get it to dissolve and form the precipitate.
I have learned that when reacting the final oxidizer and toluene, the reaction will take place in minutes rather than hours if the mixing is thorough
enough. I managed to get the last batch to fully react within about an hour at 50 C by manually mixing the two phases quite aggressively with a nickel
spatula.
Overhead stirring is definitely recommended. I don't yet have that as an option, but if I had it I would definitely use it. For a 2L size,
magnetic stirring is quite insufficient and leads to long reaction times. Strong stirring is essential for scaling up.
This evening I was going to use ether to extract out the last of the organic acid (benzoic, mostly) from the mix, however I have come home to find
that the cell is functioning just fine, several shades of pink darker that it was this morning - which is typical.
What I think I will do is make sure that ALL of the OXIDIZER is reacted before heating to drive off the excess solvent before regeneration (from now
on). I will melt up a section of glass tubing, giving one end a flat surface so I can grind up all the little chunks of oxidizer, before doing the
aldehyde reaction. That way no benzoic acid will form at any time (in theory, we shall see...).
My plan is to:
1) Let my cell regenerate as usual, should take another couple days at most.
2) Do the toluene reaction only this time make sure no oxidizer is left unreacted, accomplished by grinding up everything before doing the reaction.
3) Extract with toluene (or toluene hexane mixture) until no yellow/reddish tinge to the extract.
4) Heat it up to drive off the excess solvent.
5) Confirm that both A) no benzoic acid (or any other organic acid) was formed during the heating and that B) no oxidizable solvent is left in the
mix.
6) Electrically regenerate.
7) Repeat steps 2-6 until my 5L pail of toluene/hexane is exhausted...
If this works, then that is a perfectly workable procedure, quite easy to scale up. If not, I will try using other solvents to finish up the
extraction of the benzaldehyde and toluene, before regeneration.
In a day or two, the cell should be ready.
Then I will do another run, and post lots of pictures!
Coincidentally, my 1Kg of cinnamon oil just arrived in the post today, the smell permeating the container has made my house smell wonderful.
And, I've been trying to think up a use for my 1Kg of sodium carbonate for months, that I haven't even opened yet.
Yes indeed, if all goes well this place is going to smell like the magical cherry kingdom.
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Benzaldehyde
Toluene oxidation...
Take 3.
Okay, regeneration is nearly complete.
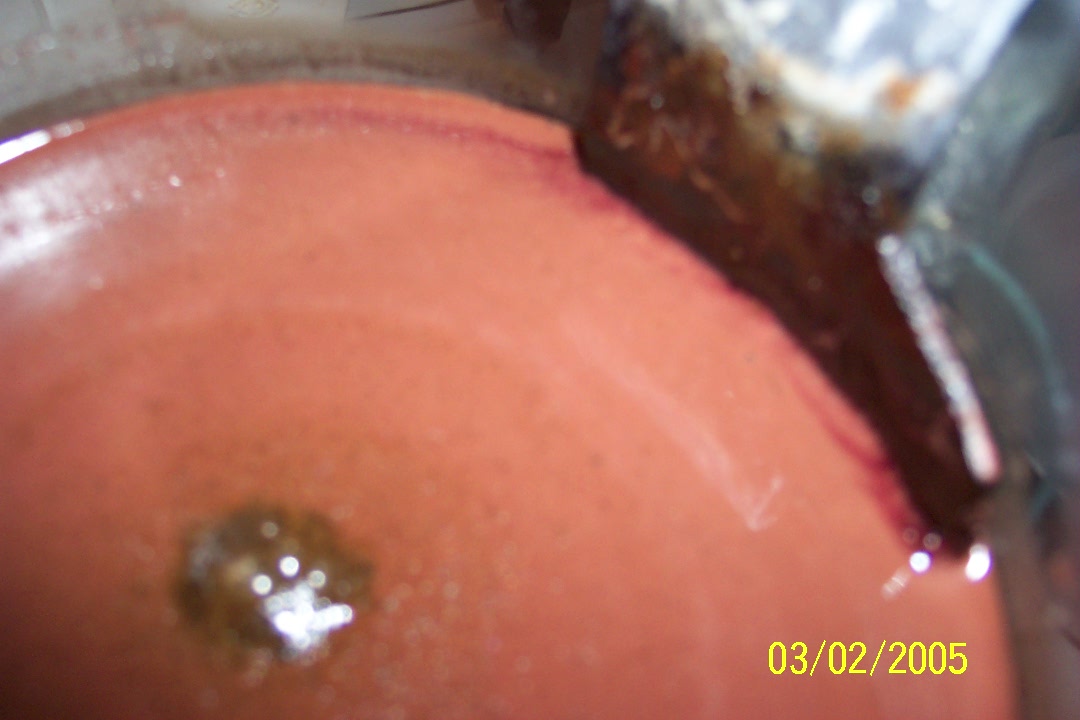
This toluene will be used for the reaction, note the considerable excess.

This will be the oxidizing mixture.

This is my glass rod I made for crunching up the little oxidizer grains.

The toluene has been mixed with the oxidizer and has been heated to 50 C, the mixture will be mixed vigorously for the next 60 minutes.

A closer look at the toluene layer...

Reaction is nearly complete.

This is the first extract, it smells very strongly of benzaldehyde. In fact, the whole house smells strongly of benzaldehyde...

All in all, I have about 80g of product, which still contains a little bit of toluene - but I won't redistill it - I'll gather it all up
with the rest and then distill it all at once.
However, my interest has been shifted in two ways, in one way I'm really fascinated by the curious oxidizing powers of the dark red salt
(manganese ammonium alum), and in another way I'm overwhelmed by the ease by which benzandehyde can be made from cinnamon oil! I'll post
those pics in just a few minutes.
Anyway, I observed that there are ways the solid oxidizer can be isolated from the mix, then reacted on its own with the toluene to form a much more
managable reaction mix. Judging by the odour generated by small amounts of oxidizer/toluene - it may even be more efficient - I don't know for
sure yet.
It is possible to use a far lesser quantity of sulfuric acid, but it means that the mixture is a paste, not a liquid. But, the paste has a much
smaller volume and hence can be "worked" in a small vessel with less labour than stirring a much larger two phase mix!
The dark red oxidizer can't be isolated on its own without decomposing, but large chunks at a time are easy to separate from the cell during
regeneration (still wet with cell acid, of course). I'm quite sure that an acidic slurry can be regenerated in a special cell (i.e. bottom of the
vessel is the anode), making the reaction much easier - and less dangerous due to the lesser quantity of acid and hence better for scaling up!
As I type, my cell is settling. That is, the manganese-ammonium sulfate is settling to the bottom. I will later pour off the top clear solution and
try to regenerate the manganese-ammonium sulfate paste in the bottom of the cell, it will be an interesting experiment.
The reason for the fascination is simple, this dark red salt produces benzadhyde on contact with toluene. Mix the two together at room temperature and
the characteristic cherry odour is noticed almost within seconds.
I can't help but think "what a handy little oxidizer.."
Heat it up and it oxidizes to the acid, keep it at 50 and it oxidizes just to the aldehyde, for toluene at least.
I'm just not crazy about the large volume of sulfuric acid, manipulations involving this cell are dangerous. The acid is concentrated enough to
dissolve cotton cloth ON CONTACT, if my 3L beaker cracked and shattered on my stove during heating, I'd hate to think of the clean up operation!
What a mess.
[Edited on 3-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Benzaldehyde from Cinnamon Oil.
Okay, this is my latest experiment, and let me say - it truly works wonders.
If you are looking for benzaldehyde, and have access to simple lab equipment, then you've got to try this one on for size.
This procedure was posted by Mendeleev and reference to the patent was given,US Patent 4,716,249.
I had been hunting this procedure for a while, but never knew how it was done. Thanks Mendeleev.
I ordered 1 Kg of cinnamon oil the same day.
This reaction converts cinnamon oil in high yields, my cinnamon oil cost me less than $30 for the Kg I ordered and contains close to 90% cinnamon
aldehyde and I've used it for this procedure without altering it in any way. This procedure only requires refluxing equipment, water, sodium
carbonate, and cinnamon aldehyde (cinnamon oil, cassia oil, cinnamaldehyde, etc).
I haven't yet isolated the benzaldehyde from the mixture, because I am running 3 batches, the third has just been started with 2 already
complete. I will separate the benzaldehyde in one single step later on, I will report back the yield, and the extracton/purification method I've
used.
As far as yields go, what I can say is that the distinctive cinnamon aroma has been replaced with a sharp, piercing benzaldehyde aroma - no mistaking
it. I think I can smell a TRACE of cinnamon, so I'm sure it must be pretty darned good!
This is the magic reagent:

And this is what 60 grams of sodium carbonate looks like:

And this is what 200g of cinnamon oil looks like:

In the oil goes:

Splash goes the oil:

Now reflux for 7+ hours:
(don't forget to use a condenser for refluxing!)


It's really simple, set up your apparatus for reluxing. I've used a 3L mantle heater set up for reflux - the thermometer is not necessary.
I've boiled 2L of WATER in a kettle. Some of which is used to dissolve 60g of SODIUM CARBONATE, the rest will be added later. Once the sodium
carbonate is dissolved, the solution is poured into the 3L 3 neck flask. Then the 200g of CINNAMON OIL is added. Then pour in the rest of the hot
water.
Add bumping granules, set up for refluxing and gently reflux for at least 7 hours. Reaction done.
Extraction of the goods should not be a problem. Reportedy, the main by-product of this reaction is cinnamon aldehyde, whcih boils at 246 C.
Benzaldhyde boils around 178 C.
From literature, the reaction should produce a mixture of 70% benzaldehyde and 30% cinnamon aldehyde.
And it sure smells like it.
I'm hoping the separation won't be difficult. I think I'll simply going to have to recommission the 'ole vigreux column for this
one. If distillation is ineffective, I will need to form the addition product to extract the benzaldehyde. I'm hoping distillation will do just
fine, though I'll probably use vacuum distillation to minimise decomposition.
After I have finished the third reaction run, I should have produced around 350 ml of benzaldehyde.
I should then have just enough recovered cinnamon aldehyde to run another batch and turn 70% of that into more goodies. To yield 400ml - 450ml
benzaldehyde, perhaps.
And it is alot easier and quicker than the acid cell method - though you can't use this procedure to make formaldehyde...
I am very pleased that this reaction actually works - and from cheap, common cinnamon oil.
Here you have it, benzaldehyde by the gallon - if that's what you want.
I'll report back the actual, final yield later on.
This reaction simply proves that things that seem too good to be true, aren't always.
Though, I hope that making benzaldehyde from cinnamon oil isn't considered "cheating..".
On one hand the chemistry is magic, but then on the other hand, perhaps making cherry flavouring from cinnamon flavouring isn't so big a deal, I
suppose...
Any comments?
[Edited on 4-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Cinnamon oil experiment update.
The cinnamon oil to benzaldehyde conversion experiment is not yet complete.
So far I have extracted and distilled all of the product from the reaction mixture.
From the 600g cinnamon oil I started with, I've managed to obtain a 500ml RBF filled to within 1 inch from the top with crude product.
The crude product is a mixture of benzaldehyde and cinnamaldehyde (and lesser quantitiesof other by-products).
What follows are the photos I've taken of the experiment so far.
This is the cinnamaldehyde fraction mixing with the benzaldehyde fraction in the 500ml receiving flask, this is the crude product.
Perhaps this photo sums up why we all love organic chemistry the best.

The photo does not do it justice. To see this process occurring in 3 dimensions with all moving "swirlies" is more information than the
human brain is designed to take in at one time. Total visual overload. I think it compares with even the best sunsets I've ever seen...Only this
is smaller - in this instance a magnifying glass achieves wonders!
And it makes a good desktop theme, at least on my 17".
I only wish I took one of higher resolution.
More swirlies..

Pure liquid glass.
And again..

Anyway, some photos of the extraction process are as follows:
Soon after the 7 hour reaction was complete.


DCM appears to be a more effective solvent than toluene for this extraction.
DCM extraction #1

DCM extraction #2

DCM extraction #3

DCM coming over...

I repeated the above, 4 times for all 6 litres. So, that's 24 extractions...That took several hours to complete. That does not include the first
extraction I did with toluene.
I would suggest to start the extractions after each reaction, rather than waiting until the end, like I did. It's alot of work and time.
However, I did have some issues with emulsions which would only break after strong HCl was added, brine alone would not break the emulsions. I had to
keep adding more solvent, until a decent separation occurred.
Have a look:
This one has just been broken and is on the retreat

(top layer is toluene)
Benzaldehyde fraction.

Distilling flask at the end of the vacuum distillation:

I am unclear whether the cinnamaldehyde is degrading or if this is the other 11% of the cinnamon oil + reaction by-products.
My diaghram vacuum pump needs overhauling, the vacuum is quite poor, the temperature in the above flask is well over 200 C.
This is why I have not carried out the final distillation of my product, it's just too valuable to risk. I will overhaul my pump, THEN do the
final vacuum distillation.
Distillation apparatus. The vigreux column was used for evaporating the solvents to minimise losses.


This is the pet store aquarium valve I use to control my vacuum distillations - it works very well. They cost about £1.20 each (couple bucks a piece)
.
They are used to regulate the air from aquarium air pumps.
I thought I would include this since I keep hearing of how "difficult" it is to regulate a vacuum during distillations. It is simple, try it
and I'm sure you will agree.
For the final benz./cinna. separation, I intend to use the column shown ealier but I will need a much stronger vacuum to do it. And I will probably
need to insulate the column to avoid excessive reflux.
Vacuum pump overhaul is next on the agenda.
[Edited on 6-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Manganese-Ammonium cell images
"You tell me man, I only work here." - Hudson, Aliens

These are what I call "anodemites".
They grow on the anode when the electrolysing solution is already saturated.
Potentially, this is a means of extracting PURE oxidizer from the cell. This is important since some cells (including mine) have other impurities
present.



Forming the oxidizing paste, without stirring.

With the pasage of more current, the red spreads to envelope the whole cell, by which time the regeneration is complete.
Another shot of the regeneration process.

The light coloured material on the electrodes is manganese-ammonium sulfate "silt" which has coated the electrodes during stirring. In time
it will all be converted to dark red manganese-ammonium alum.
What I would like to do, is make an electrode that covers the entire bottom of the vessel (anode) so that the oxidizer will form from the bottom up
into a solid red mass of crystals, which can then be removed from the acid easily, to effect toluene oxidation with minimal stirring required.
This way, I believe no stirring is required duing the regeneration. This has yet to be substantiated. I am not making THAT electrode like I did the
other 2, so I will need to actually BUY some lead sheet from somewhere, and make the bottom anode. I really want this "paste" variation to
work, since it would be so easy to scale up.
A cameras detailed impression of a "Still Experiment".

Note the headline on the newspaper, that poor guy?
[Edited on 6-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Cinnamon experiment complete
Fractional distillation of the crude product has now been completed.
The fractional distillation was carried out under vacuum, and employing a vigreux column.
The vacuum I was pulling brought the benzaldehyde over at 85 C, and the cinnamaldehyde over at 135 C.
Benzaldehyde coming over...

Cinnamaldehyde coming over...

Thermometer reads 135 C.
Column insulated for this fraction.
Benzaldehyde condensation in the still head.

Benzaldehyde collecting in receiver.

Cinnamaldehyde collecting in receiver, in it's usual, rather vibrant form.

End product.

(cinnamon left, cherry right)
Total product is :
200g benzaldehyde
128g cinnamaldehyde
The cinnamaldehyde fraction was collected until it just started to turn orange. Despite the colour, it is probably reasonably pure.
The benzaldehyde is quite pure, the suspended bubbles are droplets of solvent that came over at the very start of the vacuum distillation.
The benzaldehyde fraction was stopped when the temperature increased from 85 C to 90 C. Everything from 90 to 135 C is in the cinnamaldehyde fraction.
There is very little cinnamaldehyde in the benzaldehyde fraction, but there is bound to be a fair quantity of benzaldehyde in the cinnamon fraction
(few to several ml?).
Bumping was quite a problem during this vacuum distillation, even though the vacuum was constant, I had to stop the distillation 4 times, to add more
anti-bumping granules. I used both glass AB granules and ground pumice AB granules. This could prove to be a problem for larger batches
(distillations) when magnetic stirring is not employed. My electric heating mantles don't have magnetic stirring.
Once the cinnamaldehyde has also been converted to benzaldehyde, the total yield should be around 300g benzaldehyde.
The yield isn't as high as I rather optimistically had hoped. However, I am very pleased with the way this experiment has turned out, especially
considering that there are obvious ways the procedure can be improved.
This is the most benzaldehyde I've ever produced at one time, and from a first try!
Improvements I'm going to undertake are as follows:
1) Distill cinnamon oil before using it in the reaction, I think this may really help to avoid emulsions forming during the extraction phase. Garbage
in = garbage out. The non-cinnamaldehyde fraction of the cinnamon oil really makes a mess. The tar that is left after the distillations is completely
insoluble in spirits, soluble in toluene, but far more so in DCM, which was used to remove it from the flasks. Removing the non-cinn. fraction
(dark-brown/black) in the beginning will help to distinguish between reaction by-products and aldehyde that may be degrading at different times during
the extractions. If product is degrading, it is important to narrow down the cause.
The crude DCM extracts of the reaction mix were not washed before distillation, so some sodium carbonate will have been present - this may have
degraded a fair amount of product. The extracts were not washed because of the emulsions issue, if this is remedied by removal of the non-cinn.
fraction in the beginning, then the extracts can be washed with good separations.
2) Use magnetic stirring during the 7 hour reaction. This is based on previous experience of reacting 2 phase mixtures during reflux, yields generally
suffer markedly when proper mixing is not employed; reflux alone is not enough to break up those oily droplets.
I will need to make up an oil bath so I can use my mag. stirrer/hotplate during the 7 hour reaction.
3) Use of toluene will be avoided during the extraction. DCM will be used only.
By applying the above improvements, I think the overall yield for this procedure can be drastically improved.
[Edited on 6-2-2005 by CycloKnight]
|
|
|
chloric1
International Hazard
Posts: 1143
Registered: 8-10-2003
Location: GroupVII of the periodic table
Member Is Offline
Mood: Stoichiometrically Balanced
|
|
LOVE the picks
I understand fully about the beauty of the reaction! The pink chewed up electrode was a surreal site. Could imagine it as broken tabel leg encased
in bubble gum or a stub of a human arm in the garbage disposal. YUK! But you bring a whole new meaning to digital photgraphy! But you bring a whole new meaning to digital photgraphy!
Fellow molecular manipulator
|
|
|
Darkfire
Hazard to Others
Posts: 292
Registered: 3-1-2003
Location: California
Member Is Offline
Mood: Wondering
|
|
Cyclo this is an awsome thread and your pictures are amazing but, it would be best to link them instead of embeding them in the thread. They stretch
the page and take several minutes to load evn on dsl. This thread is likley not even loadable to people on narrrowband.
\"I love being alive and will be the best man I possibly can. I will take love wherever I find it and offer it to everyone who will take it. I
will seek knowledge from those wiser and teach those who wish to learn from me.\" Duane Allman
|
|
|
chloric1
International Hazard
Posts: 1143
Registered: 8-10-2003
Location: GroupVII of the periodic table
Member Is Offline
Mood: Stoichiometrically Balanced
|
|
Well Darkfire I ahve to agree about loading speed but these are pictures that only the finest resolution could do justice. Maybe Cycloknight could
use smaller image size with the smae pixel rating. I know sometimes I take fine resolution in small sizes to email them.
Fellow molecular manipulator
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Why not steam distill the benzaldehyde and acetal from the cinnamon oil, base mixture as it forms? Anyone have an idea how much water is needed to
steam distill 100g benzaldehyde vs 100g cinnamaldehyde?
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Benzaldehyde
Thanks for the compliments, I really enjoyed taking the pictures, I'll try to take more of future experiments.
I have a narrow band internet connection, although the first time loading the page is slow - it is quite accessible. Future visits to the page just
involves loading the new picture(s) recently added. Each image is only about 150kb average. Maybe I should have put at the top of the first page
"Narrow band users, stick the kettle on for a cuppa'. "
After all, a watched page never loads...
However, I will do something with the image size, from now on. At first I thought the pictures were expanded by this page, but it turned out to be
the other way around..
For any detailed images that are best displayed in a large format, I will simply include the scipics link to click on. That should tidy things up a
bit.
Eclectic, I believe that cinnamon oil itself is produced by steam distillation of the plant material, as are most essential oils. Steam distillation
of the reaction mixture will distill out everything except the sodium carbonate and bumping granules.
However, if pure cinnamaldehyde was used at the start of the reaction, I'm reasonably sure steam distillation could be used at the end of the
reaction to drive out the reaction products. But it would take at least a couple gallons of water (steam) to be put through each 2L quantity of
reaction mixture to get out all of the aldehyde. This would be good for processing industrial sized batches, without consuming industrial quantities
of solvents.
I may experiment with using a more concentrated solution of sodium carbonate, that way things can be scaled up greatly. I'm quite tempted to
process 1L of cinnamaldehyde in 1 reaction just to see what happens. Or maybe 500ml to start with.
The same quantity of sodium carbonate would be added, just less water.
I'm curious to see what effect that would have on overall yield.
[Edited on 7-2-2005 by CycloKnight]
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
I'm thinking that the benzaldehyde would steam distill much faster than the cinnamaldehyde. The water layer could be automatically recycled back
into the reaction mixture, and if the condenser temp was held at 30-40 C, the acetaldehyde should leave the reaction, forcing it to completion. Think
of it as an azeotrope with water. It might be worth a test run to see what king of separation you get with a mix of water, benzaldehyde, and cinnamon
oil. Also the patent recomends a surfactant, lecithin would be good for making food grade flavoring.
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Also, the sodium carbonate is just a basic catalyst for the reverse Diels-Alder addition of water to cinnamaldeyde to split it into benzaldehyde and
acetaldeyde. You could just as well start with a few tablespoons of baking soda (bicarb).
|
|
|
trilobite
Hazard to Others
Posts: 152
Registered: 25-2-2004
Location: The Palaeozoic Ocean
Member Is Offline
Mood: lonely
|
|
Retro-aldol, not Diels-Alder.
|
|
|
| Pages:
1
2
3
4
5
..
26 |
|










