| Pages:
1
..
21
22
23
24 |
elementcollector1
International Hazard

Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Also, quick question about reducing NdF3: Wasn't calcium metal suggested at one point as a suitable alternative?
HoF (CaF2) = -1228 kJ/mol (Wolfram Alpha)
2 NdF3 + 3 Ca -> 2 Nd + 3 CaF2: -370 kJ/mol, more than the equivalent with lithium. Is there some reason this is not preferred that I missed? I
read over the entire thread, but couldn't seem to find it. Is it due to calcium fluoride's high melting point?
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Update: Just got my sample of pure neodymium oxide from Snaucke Elements on EBay. It arrived as a white/gray powder, with distinct pink tinges under
incandescent light.

Most of the sample was then mixed with diluted acetic acid and boiled, in the hopes that this would dissolve the oxide. To my surprise, this worked!

Once the bubblegum-pink neodymium acetate had crashed out, I then decided to test if it dissolved in ethanol. The results gave some important clues:
-The filtrate upon first coming out was the same dark pink as the concentrated aqueous solution. This is presumably due to residual water in the
filtered acetate.
-As time went on, however, the filtered liquid became more dilute in coloration, and the drops coming out of the bottom of the filter paper were clear
in coloration. The solid acetate on top retained mostly the same volume, though it had largely disintegrated into a paste. This indicates that the
solid neodymium acetate is not soluble in ethanol, making separation of iron by selective dissolution in ethanol a viable procedure, provided the
iron/neodymium acetates are boiled to dryness.
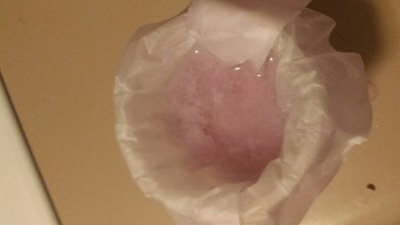
EDIT: Well, what do you know! The ethanol and water ended up separating just a few minutes after I made this post, and something cloudy has formed at
the interface.

[Edited on 12-6-2016 by elementcollector1]
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
Velzee
Hazard to Others
Posts: 381
Registered: 19-8-2015
Location: New York
Member Is Offline
Mood: Taking it easy
|
|
Yay!
Check out the ScienceMadness Wiki: http://www.sciencemadness.org/smwiki/index.php/Main_Page
"All truth passes through three stages. First, it is ridiculed. Second, it is violently opposed. Third, it is accepted as being self-evident."
—Arthur Schopenhauer
"¡Vivá Cristo Rey!"
—Saint José Sánchez del Río |
|
|
Dan Vizine
National Hazard
Posts: 628
Registered: 4-4-2014
Location: Tonawanda, New York
Member Is Offline
Mood: High Resistance
|
|
Quote: Originally posted by elementcollector1  | Also, quick question about reducing NdF3: Wasn't calcium metal suggested at one point as a suitable alternative?
HoF (CaF2) = -1228 kJ/mol (Wolfram Alpha)
2 NdF3 + 3 Ca -> 2 Nd + 3 CaF2: -370 kJ/mol, more than the equivalent with lithium. Is there some reason this is not preferred that I missed? I
read over the entire thread, but couldn't seem to find it. Is it due to calcium fluoride's high melting point? |
I'm confident that you're absolutely right and that it's superior to commercial lithium.
"All Your Children Are Poor Unfortunate Victims of Lies You Believe, a Plague Upon Your Ignorance that Keeps the Youth from the Truth They
Deserve"...F. Zappa
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
The reason lithium is preferred is its fluoride's melting point. Ideally the reaction is run similar to a thermite, where all products are in the
liquid phase. This affords good metal-slag separation.
Melting points:
Nd - 1021 C
LiF - 845 C
CaF<sub>2</sub> - 1418 C
The reaction's target minimum running temperature is one where everything is molten, thus using LiF reduces this requirement to 1021C.
The other consideration is reaction enthalpy. Careful choice of reducing agent yields extra heat as part of the reaction, helping keep your external
heating requirements low. Coincidentally I just yesterday found my calculations for reaction enthalpies for various reducing agents (the more negative
the value, the greater the heat released):
Zn - deltaH = 1020.8
Ga - deltaH = 494
Mg - deltaH = -58.6
Na - deltaH = -72.8
Li - deltaH = -193.79
Ca - deltaH = -363.73
These were calculated by using the reaction equation for each:
A NdF<sub>3</sub> + B M == C Nd + D MFx
where ABCD are the coefficients and M is the chosen metal. deltaH for elements is 0, so the calculation boils down to
dH = D*deltaH(MFx) - A*deltaH(NdF<sub>3</sub>
In retrospect, I used the values for the solids but the products will be liquid. This changes the numbers a little, but preserves their order in the
list.
Calcium produces the most heat but requires the highest temperature to melt, and I'm not sure if the tradeoff is favorable or not. If you have calcium
metal, it might be worth a shot!
I've been thinking about this experiment again recently. God, I need to just try the last step already.
|
|
|
unionised
International Hazard
Posts: 5126
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
Interesting.
"A binary eutectic composition of Formula and Formula was observed to melt at 769°C."
From
http://jes.ecsdl.org/content/104/11/661
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
Formula and Formula you say? 
From the link (in case it breaks): "A binary eutectic composition of 80.5 mole%LiF and 19.5 mole%CaF<sub>2</sub> was observed to melt at
769°C."
Somewhat less than LiF alone, but the reaction still needs to achieve 1021C to melt the Nd.
|
|
|
Lab Rat
Harmless
Posts: 9
Registered: 10-4-2018
Location: Northwestern University
Member Is Offline
Mood: Excited
|
|
This thread's been quiet for some time now... I hope my intrusion isn't too unwelcome, but I've been reading and re-reading the earlier bits of this
thread a lot in the past few weeks, and now I'm finally throwing my hat in the ring here with some questions!
I'm a simple man: all I really want is to grow some pretty pink crystals of Nd2(SO4)3*8H2O (yeah, there are other, more accessible compounds that fill
this role, but honestly I also just want the coolness of them being a Nd compound  ). Based on earlier discussions in this thread, I processed a half-inch cube Nd magnet using the oxalate separation method: demagnetize, remove
plating, dissolve in sulfuric acid, filter, oxidize all Fe(II) to Fe(III), precipitate Nd2(Ox)3 with oxalic acid. That worked fairly well, I think,
as it has for other people in this thread. Later, I was planning on converting the oxalate to the oxide, then redissolving in H2SO4 for the final
sulfate. ). Based on earlier discussions in this thread, I processed a half-inch cube Nd magnet using the oxalate separation method: demagnetize, remove
plating, dissolve in sulfuric acid, filter, oxidize all Fe(II) to Fe(III), precipitate Nd2(Ox)3 with oxalic acid. That worked fairly well, I think,
as it has for other people in this thread. Later, I was planning on converting the oxalate to the oxide, then redissolving in H2SO4 for the final
sulfate.
...Buuut, then I read Blogfast's posts on page 4, noting his discovery that Nd2(Ox)3 from this method contains significant Fe(III) contamination. 
...BUUUT, then I read Blogfast's OTHER posts on page 4 about separating Nd+3 from Fe+3 using fresh iron hydroxide/oxide-hydroxide/whatever you want to
call it! It seems perfect: FeO(OH)*nH2O is easy to prepare, and apparently, when added in excess to Nd/Fe sulfate solution, produces almost perfect
precipitation of iron as... something, the old posts are actually very unclear about what the iron becomes, but it appears that an oxide of iron is
involved.
And that's basically my question. I've literally been thinking about this iron separation problem all day, and I can't work it out. How does adding
solid Fe(OH)3 (or FeO(OH), or whatever) to a solution of a ferric salt cause all the iron cations to precipitate out? What do they turn into? What
does the Fe(OH)3 turn into? What's the reaction/mechanism? And why does it leave the Nd virtually untouched?
[Edited on 4/10/2018 by Lab Rat]
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
Doesn't look like anyone knows! Unfortunately blogfast isn't around currently to answer. Perhaps some sort of common ion effect?
I had great results with separation via the potassium sulfate double salt route. If your goal is to make crystals, though, iron removal may be less
important. The very act of growing crystals will purify your desired product! You just might have to brush the iron crud off the surface.
|
|
|
Lab Rat
Harmless
Posts: 9
Registered: 10-4-2018
Location: Northwestern University
Member Is Offline
Mood: Excited
|
|
Hey, thanks for replying! Eh, that's ok, I'll try the iron thing out on a small batch anyway just in case. And yeah, even just separating Nd salt by
crystallization is probably sufficient for me, but... I have just as much doing the chemistry as I do looking at the final product!
As for the double salt method, GAAH, it seems like everyone has had luck with that except for me! Recently, I dissolved a 45.3 gram "de-shelled"
magnet in 80 mL of 9M H2SO4, diluted it to 600 mL (to let all salts stay dissolved), filtered it, and added 7.3 g dissolved K2SO4 (which I believe is
stoichiometric)... And nothing happened. I added a bunch more, and I DID finally get precipitate, but the reaction was very slow. So okay,
whatever, chemistry isn't as clean as I dream of it being, I need to add more than one equivalent.
But the other problem is that I can tell that the reaction was incomplete, because the supernatant solution was still different colors under
fluorescent and incandescent light, indicating remaining Nd.
I figured, hey, maybe I just needed to add even more K2SO4. To test that theory before doing it, I instead made a small amount of saturated K2SO4
solution and added a few drops of my magnet solution into that. Weirdly, though, no additional precipitate!
Is it possible that my magnet solution was too acidic, causing the double salt precipitation to become unfavorable or something? Or maybe the
reaction DID go to completion, and I'm wrong about the different colors thing. When I get the chance to work on it again, I'll try oxidizing to
Fe(III) with H2O2 and testing for Nd with oxalic acid.
[Edited on 4/16/2018 by Lab Rat]
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
I ran into a similar problem. We found that just adding the solid K2SO4 directly to the magnet solution is far better
than adding a solution. I guess the extra water interferes with precipitation for some reason (doesn't make a lot of sense to me, but it does work. It
could be a pH thing like you mentioned). Just add the solid salt, stir vigorously for something like 30 minutes, allow to settle, then check the
supernatant for color. Changing solution colors definitely indicates Nd still in solution.
|
|
|
Lab Rat
Harmless
Posts: 9
Registered: 10-4-2018
Location: Northwestern University
Member Is Offline
Mood: Excited
|
|
Ooooooh, VERY good info, thanks so much! And now that I think about it, I'd be too afraid to try and modify the pH with base, because introducing
alkali or ammonium ions in the presence of sulfate might trigger early precipitation of the double salt before I want it.
|
|
|
Lab Rat
Harmless
Posts: 9
Registered: 10-4-2018
Location: Northwestern University
Member Is Offline
Mood: Excited
|
|
Ok, here's a little update from me, if anyone's interested. In my second post from a couple weeks ago, I mentioned that my double salt Nd
precipitation didn't appear to go to completion, as evidenced by different solution color under different lighting. When I got to work on it again, I
took a small amount of my solution (containing mostly Fe(ii) with suspected leftover Nd(iii) sulfates, as well as a lot of excess potassium sulfate),
filtered it, oxidized the iron, and added plenty of oxalic acid to test for neodymium. To my pleasant surprise, the test failed! No precipitate.
Either I was completely wrong before and the reaction DID go to completion and I literally just observed the color wrong, or leaving it alone for two
weeks allowed it to move towards completion. Honestly, I'm thinking it was probably the first thing lol
Filtered and washed the double salt, then reacted it with excess KOH, and filtered and washed the Nd hydroxide (pale green under fluorescent, pink
under incandescent). I'm gonna wait to continue until I finish my vacuum filter setup--all I need is the right water pump to connect to my aspirator,
and the tubing--so that the filtering and washing steps are faster.
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
Thanks for posting all this, Lab Rat! Great work!
I've been working on Nd as well. Separating it with oxalic acid worked well for me, and rinsing with ~5% H2O2 removed most of my Fe contamination. I
then thermally decomposed the Nd-oxalate, to be left with a brown oxide, the brow colour being due to contamination with Pr. A few percent of Pr is
enough to make it dark brown. When dissolving this "didymium"oxide in H2SO4 soln., the sulphate crystals were a bit more reddish than those of
completely pure Nd-sulphate.
If you decide to thermally decompose some of your Nd-oxalate, please do not strongly calcine it, or your oxide may become hard to dissolve in
sulphuric or any other acid.
Besides, I've been working on the dissolution of industrially calcined Nd2O3 (which is pure with respect to Pr-content), which is a very difficult
task. So far I failed in coming up with a good method to do this.
|
|
|
Lab Rat
Harmless
Posts: 9
Registered: 10-4-2018
Location: Northwestern University
Member Is Offline
Mood: Excited
|
|
Hey man, sounds good! Yeah, to be honest, there's no way I can expect my sample to be free of praseodymium, but the ionic radii of Pr and Nd are so
similar that I would expect Pr contamination to simply fill the same holes as Nd in the crystal structure.
As for the calcination, I've seen that issue arise once or twice in the depths of this thread, but I don't really understand it... If you heat
Nd2(Ox)3, you get Nd2O3. And if you heat it REALLY hot, you still get Nd2O3, right? How can it be that strongly calcining it makes it harder to
dissolve? Does it transform into an incredibly stable solid state structure at high temperatures or something?
As for dissolving Nd2O3, I wonder if you've seen this paper, it might be userful: https://www.degruyter.com/downloadpdf/j/ncrs.2002.217.issue-...
It's actually about dissolving praseodymium oxide to get the sulfate, but I'm pretty confident the method would work with Nd, too. Basically, they
wet the oxide powder with water first, then added conc. H2SO4 dropwise to dissolve the oxide and form the sulfate. I guess then for additional
purification/clean crystal growth, they dissolved it in 1:1 methanol:water (v/v), and added a tiny bit of 2,2'-bipyridine (presumably to form some
kind of bipy coordination complex). Evaporation yielded green needle crystals.
|
|
|
woelen
Super Administrator
Posts: 8012
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Strong heating of oxides makes the oxide more compact and more crystalline. The reactivity of the sample then strongly decreases and this can go so
far that it becomes nearly impossible to dissolve the oxide in anything else than molten glass or molten alkalies.
I have encountered many oxidis which have become like this:
- Nd2O3
- Er2O3
- Pr2O3
- SnO2
- TiO2
- Co3O4
- Cr2O3
- Al2O3
- Fe2O3
- Sb2O5
- Nb2O5
Once you have an oxide in this inert state it has become nearly inaccessible for aqueous chemistry. In this way I lost well over 100 euros worth of
money by buying oxides of metals and not being capable of using them in any meaningful way.
|
|
|
Bezaleel
Hazard to Others
Posts: 444
Registered: 28-2-2009
Member Is Offline
Mood: transitional
|
|
| Quote: Originally posted by Lab Rat | (...)
As for dissolving Nd2O3, I wonder if you've seen this paper, it might be userful: https://www.degruyter.com/downloadpdf/j/ncrs.2002.217.issue-...
It's actually about dissolving praseodymium oxide to get the sulfate, but I'm pretty confident the method would work with Nd, too. Basically, they
wet the oxide powder with water first, then added conc. H2SO4 dropwise to dissolve the oxide and form the sulfate. I guess then for additional
purification/clean crystal growth, they dissolved it in 1:1 methanol:water (v/v), and added a tiny bit of 2,2'-bipyridine (presumably to form some
kind of bipy coordination complex). Evaporation yielded green needle crystals. |
Well, it is evident from their description, that they use a weakly calcined Pr-oxide. Otherwise the dropwise addition of sulphuric acid would not make
any sense. Wetting a hard-calcined oxide does nothing at all to such oxide. (I had a bit of success by partially dissolving hard-calcined Nd2O3 in
sulphuric acid by boiling it for over an hour in a distilation set-up.)
Adding the sulphuric acid dropwise seems to me like a way of constraining the excess of sulphuric acid to a minimum. I assume that an excess of the
acid is needed in order to solve the oxide (even if it is only weakly calcined). They are still left with at least a little excess acid of though,
because they heat it to only near-dryness, instead of evaporating the excess H2SO4.
It seems to me that the addition of the CH3OH/H2O mixture is their way to neutralise the excess acid. And indeed a precipitate forms, presumably of
Pr(OH)3.nH2O. It could be that here the role of the pyridine comes in, as an agent to dissolve the hydroxide again, so that the hydroxide will not
serve as a nucleation agent for the Pr-sulphate they are after. I only suppose this; if anyone knows a better explanation, please post!
So I think that the mixture of methanol, water and the excess H2SO4 yields them a more or less neutral solution, which would prevent H2SO4 from
entering the crystal lattice. (When you crystallise Pr-sulphate from a strongly acid solution, H2SO4 *will* enter into the lattice - I know from
experience. Successive steps of calcining such results and recrystallising will reduce the amount of acid in the Pr-sulphate by a factor of ~100 in
each step.)
For me the big question about the article you mention is the role of the 2,2'-bipyridine. It is used in a large amount. The calculated molar ratio is
Pr2O3 : bipy = 1 : 1.9
or
Pr : bipy = 2 : 1.9 (almost one bipy ligand for each Pr atom)
That their solution turns orange shows that there must be a strong interaction between the bipy ligands and the f-orbitals, since the green colour of
Pr is due to transitions involving the f-electrons.
|
|
|
Lab Rat
Harmless
Posts: 9
Registered: 10-4-2018
Location: Northwestern University
Member Is Offline
Mood: Excited
|
|
Sorry, not related to what we were just talking about, but two quick things about Nd magnet separation:
1. In the sulfate double salt precipitation method, don't heat the mixture above 50°C. Honestly there's no reason to heat it at all (I don't really
know why I did it anyway), but one batch I warmed and stirred just to quicken the precipitation (which, in my experience, is a very slow reaction,
taking upwards of 24 hours to complete), and another batch I warmed upwards of 70°C and stirred, and that precipitate appears noticeably orange
(probs iron 3) under fluorescent light. I'll probably be able to separate it out somehow later.
2. I don't recommend washing/drying the sulfate double salt with acetone. I didn't have a vacuum filter setup until just now, so to dry my product,
I'd often suspend it in acetone and re-gravity-filter, because the acetone evaporates quicker than water. But in my experience, the double salt
appears slightly soluble or at least colloidal in acetone. Either way, I don't really recommend it.
Honestly, neither of these things have been suggested anywhere above, but like... just so ya know, I guess!
[Edited on 6/19/2018 by Lab Rat]
|
|
|
Dan Vizine
National Hazard
Posts: 628
Registered: 4-4-2014
Location: Tonawanda, New York
Member Is Offline
Mood: High Resistance
|
|
| Quote: Originally posted by Lab Rat |
And that's basically my question. I've literally been thinking about this iron separation problem all day, and I can't work it out. How does adding
solid Fe(OH)3 (or FeO(OH), or whatever) to a solution of a ferric salt cause all the iron cations to precipitate out? What do they turn into? What
does the Fe(OH)3 turn into? What's the reaction/mechanism? And why does it leave the Nd virtually untouched?
[Edited on 4/10/2018 by Lab Rat] |
This isn't a strictly chemical process. It has a lot to do with adsorption. Ferric oxyhydroxide (from ferric chloride plus a hydroxide) has long been
used to wash heavy metal ions out of ground-water.
It's unlikely that this method would necessarily leave Nd totally untouched, but this physisorption process is very pH sensitive and operating near
the notch of the typical V-shaped solubility curve for the targeted metal hydroxide is the aim. Normally, for ferric iron that is about pH 7.8. This
is close to the pH 7 that blogfast25 mentioned on p. 4. Presumably, the solubility minima for Nd lies at a somewhat higher pH.
"All Your Children Are Poor Unfortunate Victims of Lies You Believe, a Plague Upon Your Ignorance that Keeps the Youth from the Truth They
Deserve"...F. Zappa
|
|
|
fusso
International Hazard
Posts: 1922
Registered: 23-6-2017
Location: 4 ∥ universes ahead of you
Member Is Offline
|
|
Is there any solvents that can dissolve amorphous B? CS2?
|
|
|
Poppy
Hazard to Others
Posts: 294
Registered: 3-11-2011
Member Is Offline
Mood: † chemical zombie
|
|
Similar dissolves simular. Most like the way silicon oil cleans dust better than soap does clean it. I remind carbon is more soluble in water than
alcohol, and > than organic solvents. Thats because pi bounds and some e pairs make their fashion to surpass the dynamics of the solvents
requirements to carry the C particles along. Also, not less important you have to first of all undo the clumps of amorphic B then they will inevitably
solubilize. Then you go 2 steps: create a silly putty of B + surfactant (anything that matches the maximum dipole moment achievable for B bonds), heat
and stir, then dilute in suitable solvent for both, same way you would first rub grease with soap and they dilute both in water. For the surfactant I
would suggest sulphonic acid.
[Edited on 8-22-2018 by Poppy]
|
|
|
fusso
International Hazard
Posts: 1922
Registered: 23-6-2017
Location: 4 ∥ universes ahead of you
Member Is Offline
|
|
when I use oxalic acid to precipitate Nd3+ from the magnet chloride solution, should I add excess oxalic to complex all Fe3+ or slight excess relative
to Nd3+ is enough?
|
|
|
Lab Rat
Harmless
Posts: 9
Registered: 10-4-2018
Location: Northwestern University
Member Is Offline
Mood: Excited
|
|
| Quote: Originally posted by Dan Vizine |
This isn't a strictly chemical process. It has a lot to do with adsorption. Ferric oxyhydroxide (from ferric chloride plus a hydroxide) has long been
used to wash heavy metal ions out of ground-water. |
Ahh, very interesting, thanks a ton! I've ultimately decided that I don't trust my abilities/equipment enough to rely on this adsorbtion process, and
will instead probably just recrystallize my Nd2(SO4)3 at the end to purify. Also, I'm highly suspicious that the double salt precipitation step is pH
sensitive, and its efficiency is low in the presence of acid. I tried to use Fe(OH)3 to neutralize excess acid, but making Fe(OH)3 is a pain, and so
is filtering the excess back out (small particles). I'll try using bicarbonate in the future instead.
|
|
|
j_sum1
|
Thread Pruned
29-12-2018 at 00:18 |
j_sum1
|
Thread Split
1-1-2019 at 22:30 |
j_sum1
|
Thread Pruned
22-1-2019 at 12:52 |
j_sum1
|
Thread Split
23-1-2019 at 04:18 |
Wizzard1
Harmless
Posts: 10
Registered: 13-2-2019
Member Is Offline
|
|
To the folks getting their Nd from R.E. Magnets and HCl -
Anybody have an explanation for the perceived color differences between batches / ages of magnets? I'm assuming it's other rare-earth impurities,
mostly Pr of course.
Older magnets yield a Nd sulfate with almost a fluorescent rose color, very pink, while newer ones have a much more neutral rose color, but some
batches come out a a shade more towards lavender.
Just wondering.
[Edited on 26-2-2019 by Wizzard1]
|
|
|
fusso
International Hazard
Posts: 1922
Registered: 23-6-2017
Location: 4 ∥ universes ahead of you
Member Is Offline
|
|
I've dissolved some Nd magnets into HCl, I want to ppt Nd as oxalate. Is stoich amount of oxalic acid (just enough to ppt all Nd2ox3) enough or do I
need huge excess (much more than 3eq relative to all metals) to keep all Fe3+ as the soluble ferrioxalate ion?
|
|
|
| Pages:
1
..
21
22
23
24 |









