| Pages:
1
2
3 |
Dr.Bob
International Hazard

Posts: 2734
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
The Lee paper, along with the Wadpole JMC article, would indicate that the coupling of the azepine to the isothiocyanate is not a problem, and the
isothiocyanate can be made from the amine easily enough, so the route comes down to ways to make each part. I see few other practical ways to break
the molecule apart. Making the azepine from the amide with diborane is the way Wadpole did it, along with HBr deprotection of the dimethoxy groups,
which is not trivial, but diborane can be generated from NaBH4, so that is plausable. And HBr is not that hard to make or find. But the Lee route
to the azepine seems more doable for OTC possibilities, and the LAH reduction could be swapped for diborane also, which simplifies the chemicals to
acquire. So I would favor the Lee routes so far.
I have not seen the L Tafesse, DJ Kyle - Combinatorial chemistry & high throughput …, 2004 route yet, unless it is better, the Lee route could
be the best way to start.
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
@AVBaeyer
Because it was clear to me from the beginning that the available synthesis most probably would have been developed with a different focus than I think
is needed in order to achieve a practical OTC synthesis. So why not trust in ones own experience and creativity and discuss that here on SM. Just
grabing the literature procedure to achieve (or not to achieve) the goal by force doesn't meet my philosophy. Not to cause any misunderstandings, I'm
happy having my opinion and others having their opinion, even if this is pissing in the wind for others, although sounds cumbersome to me, no offence
intended. 
@Assured Fish
Great drawings. If I got that right the synthesis shown in the left pic changed. The butanoic acid is activated by some activating agent and then
undergoes cyclization (Friedel-Crafts acylation), then the ring is expanded by the modified Schmidt reaction.
How about the Beckmann rearrangement utilizing hydroxylamine? EDIT: The Beckmann rearrangement leads mostly to the wrong product, known at least for
the non substituted derivative.
[Edited on 9-5-2017 by Alice]
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
Alice,
No offense taken. I referenced the syntheses so that those interested in how capsazepine has been synthesized in the past could see the routes.
Gathering this sort of information is important in planning any new or modified synthesis whether starting from OTC materials or chemical catalog
materials. Designing a synthesis "de novo" without knowledge of prior work and a sound basis in organic chemistry, in my opinion, is pissing in the
wind. Organic synthesis is an excersize in reasoning by analogy. It is unusual and mostly unheard of to invent truly new reactions for specific steps
in a total synthesis. For example, look in detail at some of the great RB Woodward syntheses (eg reserpine, strychnine, vitamin B12). What you will
find is no truly new chemistry but the ingenious application of standard chemistry to new situations.
In the case of casazepine, perhaps the most satisfying excersize would be figuring out how to synthesize some of the published starting materials from
OTC sources. In a way, I think that this is the direction that this discussion may be moving which I heartily endorse. [see Dr Bob above]
I wish everyone a lot of fun in this excersize.
AvB
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Im inclined to agree with Dr.Bob, the lee ref using an intramolecular Mannich from phenylpropylamine kinda trumps most of the overly complicated
routes any of us have suggested so far.
We could still even start from safrole or methylated eugenol and hydroaminate the alkene in an anti-Markovnikov fasion however separation of the 2 and
3 phenylpropylamines may prove troublesome but i am certain their are catalysts out their that could selectively aminate at the 3 position.
| Quote: |
Great drawings. If I got that right the synthesis shown in the left pic changed. The butanoic acid is activated by some activating agent and then
undergoes cyclization (Friedel-Crafts acylation), then the ring is expanded by the modified Schmidt reaction.
|
Alice yes that has changed and also straight decarboxylation of fenconine was also suggested by CuReUS to go straight to the amine.
The only thing is that most decarboxylations of 3-amino-3-ethanoic acids are a pain in the ass to pull off e.g. tryptophan.
TCCA is OTC as fuck and a nitrile reduction to an amine is not all that difficult to achieve if you have hydrides on hand, LAH i know we are trying to
avoid but the nickel boride reduction using NaBH4 and NiCL2 is quite straight forward and should get quite good yields.
I am still very confused as to how CuReUS's azepine ring cylization works though, you mention using TCAA i suspect trichloroacetic acid.
| Quote: |
In the 2nd step,the acid undergoes an intramolecular FC acylation using TCAA to form the ketone
|
CuReUS could you elaborate further or perhaps draw it out for us pretty please?
|
|
|
clearly_not_atara
International Hazard
Posts: 2788
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
| Quote: | | the lee ref using an intramolecular Mannich from phenylpropylamine kinda trumps most of the overly complicated routes any of us have suggested so far.
|
Hey I also used an intramolecular Mannich... although I severely underestimated the difficulty of reducing Cp2ZrCl2, which it turns out can pretty
much only be done with aluminium hydride reagents. The best route I found so far uses DIBAL, which can be made OTC (or some similar replacements) but
is well worth avoiding.
I think that you could actually get away with performing the radical addition of HBr to safrole. As long as the conditions are nonpolar enough, Br-
ions won't be present, and the ether won't be protonated, which means it won't be cleaved. Probably benzene is the best solvent since I'm not sure how
well safrole will dissolve in hexane. I'm also not sure what the best radical initiator is, but you can make something like AiBN by making butanone
azine the usual way and converting the hydrazones to tetrahedral carbon atoms with a carbanion like ethyl acetoacetate. Or tert-butyl peroxide might
be accessible somehow but I have no idea. I think methyl iodide and ultraviolet light counts as a radical initiator in some jurisdictions.
From there you can staple on the amine of your choice or potassium isocyanate if for some reason you actually want to use isocyanates like some people
in this thread.
Actually it's possible that if you try to perform the Delepine on the benzodioxol-5-propyl bromide you'll end up forming the azepane because the
transient iminium will attack the arene rather than being hydrolysed by water. It might even be possible to "force" this reaction by decomposing the
hexamine onium with acid in the absence of water. This would be a particularly elegant approach if it worked:
EDIT: LOL there's an extra "N" in there somewhere, stupid drawing software

[Edited on 10-5-2017 by clearly_not_atara]
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Here is another draft, applying some changes, especially four points mentioned by clearly_not_atara, which is avoiding K, avoiding ether at least in
one step, acetal protection, and applying the Delépine reaction.
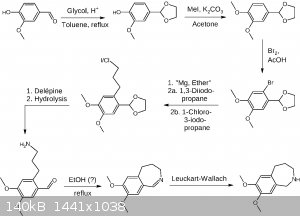
EDIT: Revised route.
Not sure about the Leuckart-Wallach and maybe the formaldehyde formed in the Delépine reaction may lead to some unwanted side reactions, which may be
circumvented by using the Gabriel synthesis or succinimide as the amine donor. EDIT: Buffering the bromination may be neccessary in order to save the
acetal from deprotection.
[Edited on 10-5-2017 by Alice]
@Assured Fish
| Quote: | Alice yes that has changed and also straight decarboxylation of fenconine was also suggested by CuReUS to go straight to the amine.
The only thing is that most decarboxylations of 3-amino-3-ethanoic acids are a pain in the ass to pull off e.g. tryptophan. |
I think phenylalanine is relatively easy to decarboxylate. The problem with tryptophane is the indole moiety.
| Quote: | | TCCA is OTC as fuck and a nitrile reduction to an amine is not all that difficult to achieve if you have hydrides on hand, LAH i know we are trying to
avoid but the nickel boride reduction using NaBH4 and NiCL2 is quite straight forward and should get quite good yields. |
As you say, 'if at hand'. For me NaBH4 falls in the same category as LAH although its reactivity is weaker. Non OTC chemicals in my opinion. Diborane
as a substitute has an autoignition temperature of 38 °C. Boranes are great rocket propellants.
| Quote: | | I am still very confused as to how CuReUS's azepine ring cylization works though, you mention using TCAA i suspect trichloroacetic acid.
|
It's the anhydride which leads to a mixed anhydride with the butanoic acid which in return is easy to cyclisize. In the mixed anhydride the
trichloroacetyl part draws charge from the butanoic part making it very susceptible for the arene. Additionally trichloroacetate is a very good
leaving group. There are loads of procedures published for alternate F-C-acylations. It's also possible cyclisizing acids directly, but the conditions
are usually harsh. Maybe some milder conditions would work here as the attack happpens para to MDO.
[Edited on 10-5-2017 by Alice]
[Edited on 10-5-2017 by Alice]
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Lee & Lee report, that the intramolecular Mannich reaction proceeds very sluggishly and yields not the right product as main product, but the
N-methylated derivative. Their key finding is N-benzylation and a free phenol para to where the Mannich should happen. This means, the product of the
reaction is benzylated and in a next step cleaved by H2 baloon and Pd/C. On the other hand I guess if both methoxy groups are cleaved beforehand the
Mannich also happens in ortho-phenol position. If none are cleaved the reaction might also not give such good results.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Dr.Bob  | But the Lee route to the azepine seems more doable for OTC possibilities,So I would favor the Lee
routes so far. the Lee route could be the best way to start. |
I respectfully disagree.I don't think either trimethylphosphoacetate or isovanilin is available OTC and neither is
1,1'-thiocarbonyldi-2-(1H)-pyridone)
You know how to make hydrides at home ? 
| Quote: Originally posted by Assured Fish | | Im inclined to agree with Dr.Bob, the lee ref using an intramolecular Mannich from phenylpropylamine kinda trumps most of the overly complicated
routes any of us have suggested so far. |
But keep in mind that the amine has to be protected and deprotected,like alice said above,which makes the reaction redundant
| Quote: | | We could still even start from safrole or methylated eugenol... |
Not if we go via the mannich.Presence of a free para OH is essential for the reaction.Read the above post by alice.
| Quote: | | The only thing is that most decarboxylations of 3-amino-3-ethanoic acids are a pain in the ass to pull off e.g. tryptophan. |
I am sorry,I should have given you a ref related to phenylalanine(table 2,entry 7).There is a slight misprint,the decarboxylation gives
b-phenethylamine only,not alpha-https://erowid.org/archive/rhodium/chemistry/trp.decarbox.en...
| Quote: | | I am still very confused as to how CuReUS's azepine ring cylization works though, you mention using TCAA i suspect
trichloroacetic acid. |
That's where you are making a mistake.The cyclisation won't give you the azepine directly,it will only give the ketone.You will have to then convert
the ketone in 2 steps(modified schimdt followed by reduction) to the azepine.
I am attaching a pic of the route.As for the cyclisation mechanism,Alice has hit the nail on the head and explained it beautifully.
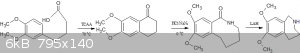
| Quote: Originally posted by Alice | | On the other hand I guess if both methoxy groups are cleaved beforehand the Mannich also happens in ortho-phenol position. |
Not to mention the crap formed by the reaction of HCHO with the diol
[Edited on 10-5-2017 by CuReUS]
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Here is my third draft, reintroducing the yeast mediated reduction of vanillin and K for its global deprotonation. Not sure but would be fortunate if
KOH works instead. A method for the extraction of vanillyl alcohol is required, which doesn't end up in a mess and too much chemistry in buckets. EtI is introduced in order to avoid MeI. The second last step may be omitted which
may reduce the synthesis to 7 steps (not counting reagent prearations). Although I suspect the resulting product will have a higher purity going for
the quarternary ammonium salt, hopefully making intermolecular reactions especially between the amine and the benzyl bromide less likely.
Trimethylamine may be obtained by treatment of any available quaternary trimethyl ammonium salt with a strong base like KOH. In this synthesis a
global deprotection (hopefully leading to the benzyl bromide intermediate) was chosen, avoiding an extra step for the aryl ether deprotection. I would
not recommend isolating the bifunctional amine/bromide, just boiling off the HBr, adding it to a new solvent containing trimethylamine. Something
which wasn't discussed before but I think is imporant to know is the deprotection utilizing HBr will yield bromoalkyls, so be aware of the gases
formed.
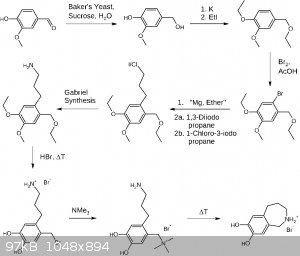
| Quote: | | As for the cyclisation mechanism,Alice has hit the nail on the head and explained it beautifully. |
Thanks!
| Quote: | | Not to mention the crap formed by the reaction of HCHO with the diol
|
Different types of resin.
[Edited on 11-5-2017 by Alice]
[Edited on 11-5-2017 by Alice]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
You could have done the synthesis in 6 steps by hydrolysing the ethers and converting the benzyl alcohol to bromide in the 2nd step(acid hydrolysis)
of the gabriel itself.But you would have to free base the bromide salt before the cyclisation,which would add one more step to make the synthesis 7
steps.
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
I thought about this. The reason I decided not to suggest it is a possible activation of the carboxylic groups by HBr with possible Friedel-Crafts
products. This may be even a bigger problem as the phenols are deprotected then. If it turns out there is no such problem there is indeed a step to
omit.
I think free basing is a very very critical step as there is no good reason why there would be no excessive chain formation. Would be a pitty losing
lots of product in the last step and I don't know how the product may be separated other than a column. Therefor catching the benzyl bromide by
trimethylamine may be the safer way, not just because the reaction is probably faster than either chain formation or cyclization but to additionally
accomplish the following cyclization as controlled and clean as possible. This may be accomplished by preparing a hot solvent and to introduce the
previously diluted quarternary ammonium compound as slowly as possible.
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
I have been following this thread with considerable interest, although for the most part it is a little over my head. I find the notion that baker's
yeast can do reductions to be fascinating. Is this the same common bread/brewing/wild yeast that can be found almost everywhere or is it a special
strain? Also, can it reduce aldehydes generally?
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
@JJay, the brand mentioned in the paper is Fleischmann's "active dry" yeast. I don't know this particular brand but it looks like usual baker's yeast.
There are lots of publications focusing on yeast mediated reducion of ketones especially aryl alkyl ketones. I didn't know before it works for
vanillin too. I didn't find much about aldehydes. If you are interested, did you know carrots can be used as well for ketone reductions into chiral alcohols?
That might be interesting for vanillin reduction too.
[Edited on 11-5-2017 by Alice]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
Wow, that is pretty much the most garden variety of yeast I can think of. I actually have a stray packet of it sitting around here somewhere. Carrots,
LoL. That is interesting, though....
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: | | I think free basing is a very very critical step as there is no good reason why there would be no excessive chain formation. |
But if you don't free base it,the NH2-Br salt won't react with the benzyl bromide.Also,I just remembered that catechols get oxidised very
fast in the presence of base,so the free-basing step is a necessary evil
If you think of it,its nothing great actually.Yeast has been used for thousands of years for fermentation(breaking down sugars to acetaldehyde and
then reducing it to ethanol).So its obvious that it can also be used to reduce other substrates.Read this review -http://pubs.acs.org/doi/abs/10.1021/cr00001a004 | Quote: Originally posted by JJay | | Wow, that is pretty much the most garden variety of yeast I can think of. I actually have a stray packet of it sitting around here somewhere.
|
if you are planning to do this reaction,keep in mind that you will need a centrifuge(according to the vanillin reduction paper),probably to separate
out the yeast cells finely suspended throughout the mixture before you can get your product.
[Edited on 12-5-2017 by CuReUS]
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
| Quote: |
But if you don't free base it,the NH2-Br salt won't react with the benzyl bromide.Also,I just remembered that catechols get oxidised very fast in the
presence of base,so the free-basing step is a necessary evil
|
It is possible for us to freebase or demethylate a quarternary ammonium salt without using strong bases which might react with the catechol.
https://www.thevespiary.org/rhodium/Rhodium/Vespiary/talk/in...
from what i understand they suggested using a neucleophilic amine that is more neucliophilic than the ammonium salt they wish to freebase to which the
alkyl halide would prefer, they suggested using ethanolamine and cited a reference to go with it.
|
|
|
Sigmatropic
Hazard to Others
Posts: 307
Registered: 29-1-2017
Member Is Offline
Mood: No Mood
|
|
I’ve seen this thread pop up more than once in today’s posts over the past weeks and have given the idea some thought.
First off I will go through this without any of the OTC fetish and establish the general scheme. Later I will comment on certain steps and suggest
possible OTC alternatives. If you have any additional suggestions, blast away!
So I´d start from catechol and suitably protect this with some alkyl groups. Compound 1 would be then be subjected to Friedel-Crafts acylation with
succinic anhydride. After separating the other possible isomer of 2, 2 would be subjected to a Curtius rearrangement in which the heating step would
be performed in an anhydrous environment. This would hopefully give the isocyanate, which upon treatment with anhydrous acid would cyclisize to form
3. Reduction/deoxygenation under harsh conditions should furnish 4 which would be deprotected to 5 by refluxing in HBr.
Then onto the p-chlorophenethylamine isothiocyanate. Ring chlorination, radical benzylic chlorination and substitution with a cyanide salt as per
NurdRage. Then reduce 8 with alane (fearing loss of the ring Cl when using straight LAH). Activation of 9 with thiophosgene should allow it to be
attached to 5 and yield capsazepine.
Overall I think it is pretty atom efficient except for the steps where isomers can be formed: 12, 23 and PhMe6. I've looked in reaxys and
most reaction are known, although not for the particular compounds. Having said that I'm sure I could provide credible references on most reactions if
It weren't for me closing all the tabs and discarding the information last night.
The main problems for OTC style would be Curtius&cyclization 23, reduction 34, reduction 89 and 910.
23: instead of using DPPA or SOCl2 then NaN3 one could use TCCA to make the acid chloride and then make the acyl azide. Isolate that and heat it in
anhydrous conditions to give the isocyanate.
34: Somewhat more problematic but possibly HI/Red P could work, although I have my doubts about reduction of the amide. These conditions may even
do a tandem demethylation making it 35
89: May be achieved by aluminium amalgam reduction. There are plenty of routes to phenethylamines.
910: The reason why I think this is not an OTC style synth. and it is not worth bothering starting from absolute schratch. Although conceivably it
is possible from C/SCS2 + Cl2 CSCl2
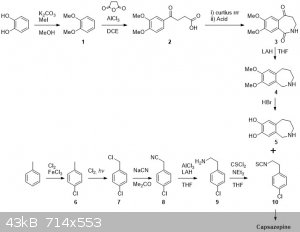
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
CuReUS, I think boiling 48% HBr isn't exactly a very good hydrolytic condition. In the following I show you what I'm afraid might happen.
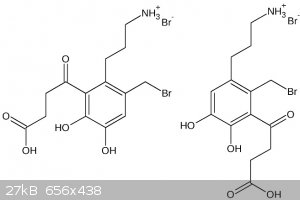
| Quote: | | But if you don't free base it,the NH2-Br salt won't react with the benzyl bromide.Also,I just remembered that catechols get oxidised very fast in the
presence of base,so the free-basing step is a necessary evil
|
I didn't mean not to free base the amine at all, just that it won't be trivial, because chain formation will start instantly. I'm not familiar with
the oxidation of catechol especially kinetics. If nothing else helps then intert gas (N2 balloon) is the answer.
In the meantime I checked the pKb in water for trimethylamine and butylamine (as a rough comparison), it's 4.19 for the former and 3.22 for the
latter. This opens the door for not massively liberating the propylamine while adding trimethylamine, at least in equilibrium. In reality the
interconversion happens fast but the reaction of equimolar amounts leads to an equilibrium constant of around 10:
propylammonium bromide + trimethylamine <=====> propylamine + trimethylammonium bromide
So the acid base reaction is 10 times faster from the right to the left than the other way round. Adding an equimolar amount dilute trimethylamine
into the benzyl bromide (the propylamine still protonated) solution very slowly might be better than reverse.
@Assured Fish
| Quote: | | It is possible for us to freebase or demethylate a quarternary ammonium salt without using strong bases which might react with the catechol.
|
Why would you want to demethylate the quarternary ammonium salt? Free basing would mean to remove a proton from primary, secondary or tertiary amines
ammonium salts. As the benzyltrimethylammonium salt doesn't have a proton attached to nitrogen (as it's quaternary) there is no possibility for free
basing. If any group is gonna be displaced from the quaternary salt by a good nucleophile it's the trimethylamine part which is the leaving group
then. But this is exactly my intention - reacting the free based propylamine in an intramolecular Sn2 reaction with the quarternary benzylammonium
bromide.
Free basing in this context means liberating the propylamine from propylammonium bromide by neutralizing "HBr" with a base, which otherwise blocks the
amine so it can't act as a nucleophile. In the protonated form there is no lone pair which could do that.
[Edited on 12-5-2017 by Alice]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Assured Fish |
It is possible for us to freebase a quarternary ammonium salt without using strong bases which might react with the catechol. |
The salt I am referring to here is a primary amine salt,not a quat
both the refs I found on FC acylation using isocyanate use BF3.Et2O
https://en.wikipedia.org/wiki/Curtius_rearrangement#Dievodia...
http://pubs.rsc.org/en/Content/ArticleLanding/2014/QO/c4qo00...
| Quote: Originally posted by Sigmatropic | | 34: Somewhat more problematic but possibly HI/Red P could work, although I have my doubts about reduction of the amide. |
HI/red P would break the lactam
| Quote: Originally posted by Alice | | CuReUS, I think boiling 48% HBr isn't exactly a very good hydrolytic condition. In the following I show you what I'm afraid might happen.
|
I think the succinic acid would react with the diol rather than undergoing a FC acylation.
| Quote: | | Free basing in this context means liberating the propylamine from propylammonium bromide by neutralizing "HBr" with a base, which otherwise blocks the
amine so it can't act as a nucleophile. In the protonated form there is no lone pair which could do that. |
what other context is there for free basing ? That's what it means in all contexts.
[Edited on 13-5-2017 by CuReUS]
|
|
|
Eddygp
National Hazard
Posts: 858
Registered: 31-3-2012
Location: University of York, UK
Member Is Offline
Mood: Organometallic
|
|
OK, as a small incentive*, I'll offer ~2.5 g or so of fairly pure brominated cinnamic acid (synthesised by yours truly) and 0.5 g or so of very pure
p-nitrophenyl benzoate to whoever manages to design a home-chemist-friendly route to capsazepine. By the 20th of June.
* Europe guaranteed; other continents... if you could enlighten me on shipping etc. that would help
[Edited on 14-5-2017 by Eddygp]
there may be bugs in gfind
[ˌɛdidʒiˈpiː] IPA pronunciation for my Username |
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
@CuReUS
EDIT:
| Quote: | | if you are planning to do this reaction,keep in mind that you will need a centrifuge(according to the vanillin reduction paper),probably to separate
out the yeast cells finely suspended throughout the mixture before you can get your product. |
Of course the procedure I found was an educational example on very small scale, it doesn't mean it's always the case removing yeast by a centrifudge.
As I think the fermentation of vanillin is very interesting, I finally started doing a bit of literature research about how to remove the yeast other
than by a centrifudge. I found loads of information about filtering techniques, so I think I will give it a try, as this might be interesting in
general not just for the capsazepine synthesis.
| Quote: | | I think the succinic acid would react with the diol rather than undergoing a FC acylation. |
Both isn't completely impossible, so I wouldn't exclude any possibility. Especially the esters may be activated by HBr and undergo acylation. If
esters form, although it's reversible, it wouldn't solve the goal omitting a step, especially a polyester would be unfortunate.
| Quote: | | what other context is there for free basing ? That's what it means in all contexts. |
I have explained what free basing means in general just in the previous paragraph. Context in this context means the molecule we're talking about. But
I'm pretty sure you know how it was meant.
@AvB. Sorry for the late answer, I was busy. You are speaking very indirect and general, so I have no clue what you're talking about. Do you think I
don't have a sound understanding of organic chemistry or do you think someone having no sound understanding of organic chemistry should do this and
that instead of thinking about new total synthesis? Do you think I don't know anything about total synthesis or are you explaining total synthesis in
general? Do you think I should utilize standard reactions or do you think others should utilize standard reactions? How would you define standard
reactions and what would be non standard reactions then? If this happens to be a controversy, I'd like to know what's your point.
| Quote: | | In the case of casazepine, perhaps the most satisfying excersize would be figuring out how to synthesize some of the published starting materials from
OTC sources. |
I took a different path - as a conscious decision. Anyone can choose whatever exercize this may be. You named another one, more beginner friendly
perhaps. As I tried to explain you before, you can either choose to develop OTC reagent synthesis for known routes or you can develop a route
according to OTC reagent availability. Both has it's own charm, but there is no such thing like right and false here.
[Edited on 14-5-2017 by Alice]
[Edited on 14-5-2017 by Alice]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
No I didn't,I swear.In fact,I feel like an idiot now for asking such a stupid question
| Quote: | | Both isn't completely impossible, so I wouldn't exclude any possibility. Especially the esters may be activated by HBr and undergo acylation. If
esters form, although it's reversible, it wouldn't solve the goal omitting a step, especially a polyester would be unfortunate. |
yeah,you are right.8 steps it will have to be
btw,I just noticed something funny.In the reaxys pdf posted by waffleSS,the benzazepine is first converted to the bromide salt before reacting it with
the isothiocyanate.Why can't you just react it directly ?
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
I honestly thought you were just kidding me.
| Quote: | | yeah,you are right.8 steps it will have to be |
Possibly adding sodium succinimide after the grignard one-pot would safe a work-up.
| Quote: | | btw,I just noticed something funny.In the reaxys pdf posted by waffleSS,the benzazepine is first converted to the bromide salt before reacting it with
the isothiocyanate.Why can't you just react it directly ? |
I think this has just something to do with the demethylation by HBr done before. I checked one paper (page 9) and in the following step, there is triethylamine added. So it has no impact on the reaction, the amine will just be stable for
storage.
[Edited on 14-5-2017 by Alice]
[Edited on 14-5-2017 by Alice]
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Sorry i misunderstood what alice wanted to do with that reaction.
I must admit you guys tend to loose me alot in this thread, i feel like the fumbling bafoon trodding along after you guys with pockets full of stones
| Quote: |
OK, as a small incentive*, I'll offer ~2.5 g or so of fairly pure brominated cinnamic acid (synthesised by yours truly) and 0.5 g or so of very pure
p-nitrophenyl benzoate to whoever manages to design a home-chemist-friendly route to capsazepine. By the 20th of June.
|
Exactly how "home-chemist-friendly" are we talking about here?
Because a lot of the routes suggested already are pretty damn close, with only a few issues arrising by the use of some niche reagents such as
trichloroacetic anhydride, Diisobutylaluminium hydride, sulfides etc.
These reagents are difficult to synthesize for sure but could be achieved by a determined amateur chemist.
Also are hydrides and carbon disulfide sufficiently "home-chemist-friendly" for your liking? Hydrides can after all be bought from other members on
the forum, although CS2 is a bitch to make.
[Edited on 15-5-2017 by Assured Fish]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
isn't there a chance that some of the unreacted dihalopropane might also react with the succinimide ?
just buy it,for chemistry's sake
|
|
|
| Pages:
1
2
3 |








