| Pages:
1
2 |
Darkstar
Hazard to Others

Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
Quote: Originally posted by Magpie  | | So, I will try this synthesis again. I will be trying to avoid conditions such as overheating that would promote oxidation of the 2-octanol.
|
I think your main culprit was just lack of 2-octanone reduction, not so much 2-octanol oxidation. In fact, I wouldn't be surprised if you never
actually produced 2-octanol to begin with. When you attempt the synthesis again, try to stick as closely to the procedure as possible. Like
clearly_not_atara said, it's generally not a good idea to make alterations to a known procedure unless you're 100% sure you know exactly how the
reaction works. The lack of water return may very well have been one of the key reasons your synthesis failed. You must assume that everything in the
procedure was done for a reason. And as I demonstrated in my mechanism, water does take part in the reaction. It is necessary for both the
formation of the aldol, as well as the protonation of the enolate to give 2-octanone.
Also, as suggested by Dr.Bob, conducting the reaction in a more tightly sealed vessel next time might also give better results. As I've mentioned
numerous times, the reduction of 2-octanone by H2 is how the 2-octanol is produced. I can't stress enough how critical this part of the
reaction is. Without this step, there can be no 2-octanol in the first place! Try to keep the 2-octanone in contact with the hydrogen gas for as long
as possible. This is also why it's probably a good idea to extend the reaction time for as long as possible as well.
Ask yourself this: if the pyroysis of ricinoleic acid went to completion for both of you, how is it that the Vasishtha reference somehow got a
higher yield of 2-octanol than you, but a lower yield of sebacic acid? The most likely answer? Their reaction time was more than
twice as long as yours. This means their 2-octanone had much more time to be reduced to 2-octanol by H2, and, consequently, some of their
10-oxodecanoic acid to be reduced to 10-hydroxydecanoic acid as well. (hence the lower yield of sebacic acid)
And lastly, if you have any more 2-octanone leftover from your first attempt, maybe add it to the reaction vessel next time as well. More 2-octanone
around means more opportunities to be reduced to 2-octanol and thus better yields. Even though in theory there's only enough H2 to reduce
just the 2-octanone generated in that specific reaction since they react in a 1:1 molar ratio, in practice the reduction will be far from 100%
efficient, and there will inevitably be a lot of H2 that ends up evolving as hydrogen gas. So hopefully all that extra 2-octanone can react
with some of the H2 that would have otherwise been wasted.
[Edited on 11-14-2015 by Darkstar]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
In this procedure 2-octanone is reduced to 2-octanol via Meerwein-Ponndorf-Verley reduction (alcoholate + alcohol), AFAIK. There's no way hydrogen can
become active in the reaction.
|
|
|
AvBaeyer
National Hazard
Posts: 654
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
I agree with byko3y. Once H2 is formed it is useless in this reaction. There appears to be no way that it can be "reactivated" as a reducing agent,
particularly since the reaction is vented. The reduction must occur through some sort of hydride transfer such as the M-P-V reaction that byko3y
mentions or some other sort of hydride shift. Perhaps the red lead is playing a redox role by shifting between oxidation levels and facilitating the
reduction via electron transfer.
AvB
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
I am wondering if it's possible that H2 activation could be affected by the copper vessel somehow? Perhaps in the manner that Dr. Bob
suggested? I have no problem accepting an MPV-style reduction involving a hydride shift, but I'm still curious as to why Magpie didn't get any
2-octanol in his first attempt.
[Edited on 11-15-2015 by Darkstar]
|
|
|
Dr.Bob
International Hazard
Posts: 2766
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
Perhaps adding some isopropanol to the reaction might help drive the reaction then. I was figuring that any hydrogen reduction had to be using the
lead as the transition metal, which is likely reduced and would then act catalytically on the ketone. I agree that hydrogen itself won't likely
reduce the ketone just by itself in that situation. The need for lead and a copper vessel suggests that you might even make some small amount of a
more complex metal complex in situ. I was just trying to think of ways to help the reaction. Would adding a small amount of copper dust allow the
reaction to proceed in a glass vessel?
|
|
|
AvBaeyer
National Hazard
Posts: 654
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
Here is a recent abstract though it also invokes microwave chemistry, for what it is worth:
Obtaining 2-Octanol, 2-Octanone, and Sebacic Acid from Castor Oil by Microwave-Induced Alkali Fusion
Nezihe Azcan * and Elif Demirel
Anadolu University, Faculty of Engineering and Architecture, Department of Chemical Engineering, 26470 Eskisehir, Turkey
Ind. Eng. Chem. Res., 2008, 47 (6), pp 1774–1778
DOI: 10.1021/ie071345u
Publication Date (Web): February 21, 2008
Copyright © 2008 American Chemical Society
Abstract
In this study, the effect of microwave irradiation on alkali fusion of castor oil was investigated using different NaOH/oil ratios (8:15, 12:15, and
14:15), reaction temperatures (473, 493, 503, 513, and 523 K), and reaction times (15, 20, 25, and 30 min) in order to obtain oleochemicals
(2-octanol, 2-octanone, and sebacic acid) in the presence of 1% catalyst (Pb3O4) by weight (w/w). The yields of 2-octanol (4.4−62.6%), 2-octanone
(1.6−37.4%), and sebacic acid (9.1−76.2%) were obtained according to reaction conditions. Maximum yields of oleochemicals were obtained using
methylated then presaponified castor oil (sodium ricinoleate), 14/15 NaOH/oil ratio at temperature 513 K and 20 min reaction time. 2-Octanol,
2-octanone, and sebacic acid were determined by GC and GC/MS analysis, and purity of sebacic acid (98.7%) was further assessed by its acid value (444)
and melting point (406.5 K). Reaction time compared to conventional heating was dramatically decreased by microwave heating system from 5 h reaction
time to 20 min.
It appears that the mix of products is condition dependent but we would need to see the actual data to evaluate same. I would hazard a guess that
microwave conditions would produce the same product profile as regular thermal conditions.
AvB
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
I had picked up that paper at the library yesterday, AvB, but had forgotten to look at it - thanks for the reminder. Here it is:
Attachment: 2-octanol & sebacic acid from castor oil by microwave.pdf (194kB)
This file has been downloaded 516 times
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
AvBaeyer
National Hazard
Posts: 654
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
Magpie,
Thanks for the download. I will take a look.
AvB
|
|
|
gsd
National Hazard
Posts: 847
Registered: 18-8-2005
Member Is Offline
Mood: No Mood
|
|
Magpie,
This paper could be of some help to you for whatever it is worth.
gsd
Attachment: Chemical derivatives of castor oil.pdf (743kB)
This file has been downloaded 553 times
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thanks gsd. That is a very comprehensive coverage of a much researched raw material.
I'm glad I was never required to drink castor oil!
I always wondered what the Castrol motor oil was all about. I read that it contains castor oil which at the time it was invented provided superior
lubricating properties for auto racing.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
The Volatile Chemist
International Hazard
Posts: 1981
Registered: 22-3-2014
Location: 'Stil' in the lab...
Member Is Offline
Mood: Copious
|
|
Nice article. Will make good reading tonight 
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
After learning in the above experiments that I’ve been making 2-octanone rather than 2-octanol I began trying to improve my use of the Vasishtha
procedure.
Batch # 6 (1/6 scale). This attempt added distillate water recycling to the pyrolysis pot, as did Vasishtha. This produced a large
volume of cloudy distillate: ~45 ml. It did clear up when dried with K2CO3 overnight. However, when I attempted to fractionally distill it using a
Vigreux column I had to abort due to severe foaming. I suspected that the pyrolysis distillate was contaminated with light components from the
mineral oil.
Vacuum Distillation of Mineral Oil. Convinced that my mineral oil was too light (Vasishtha specified a bp of at least 380°C) I
attempted to vacuum distill my oil at 1mmHg, discarding anything coming over at <198°C. Due to severe bumping, even with an ebulliator tube, this
was aborted.
Batch #7 (1/6 scale). Paraffin wax is a mixture of alkanes with bp >350°C. Therefore I attempted to substitute paraffin wax for
mineral oil in the Vasishtha procedure, again with distillate water recycle. But bits of wax were forming in the condenser so this attempt was also
aborted.
At this juncture I decided to switch from the Vasishtha procedure to the OrgSyn (B) procedure.
Batch #8 (0.011 scale). The OrgSyn (B) procedure also uses distillate water recycle. Again I used my copper 500ml RBF as pot. The
distillate was captured using a downward sloping condenser. This distillate was slowly reintroduced to the pot using a P-E funnel located in the
short neck of a Claisen adapter. Not too long into the pyrolysis severe foaming overflowed and plugged the condenser. This attempt was aborted.
Batch #9 (0.0055 scale). OrgSyn cautions that there must be sufficient freeboard in the pot due to foaming. Therefore the batch
size from above was cut in half. The quantities used were: castor oil, 65.1g; NaOH, 16g; water 32 ml. The components were mixed to saponify the oil
prior to transfer to the pot. I recommend that the transfer be made before the oil turns into the hard curd shown below.

saponified castor oil curd
The same distillate water recycle system was used as in batch #8, as shown in the pictures below.

OrgSyn (B) apparatus

distillate water recycle apparatus
Once 10 ml of water had accumulated in the separatory funnel it was drawn off and transferred to the P-E funnel. From there it was reintroduced to
the pot drop-by-drop. There was no foaming problem. The electric mantle heat was set at 65% with the pot and Claisen insulated with fiberglass
blankets. Stirring was done using a magnetic stirrer. The putative 2-octanol (oil) and water co-distilled, separating in the condenser. After
setting overnight in the separatory funnel the oil and water layers separated nicely as shown in the picture below.

distillate receiver
The yield was ~ 9 ml of crude oil, which is about a 26% yield.
The procedure for batch #9 will be repeated until at least 40 ml of oil have been acquired. Then I will fractionally distill it to hopefully obtain
the pure 2-octanol.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Very good!
Btw. isn't 2-octanone a useful compound? If you made it rather than the 2-octanol at first then that procedure might also be worth optimizing.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thank you. Yes, the 2-octanone and sebacic acid have been saved for possible future use. At this time I will leave the optimization to others.  Such research can be very time and material intensive. Such research can be very time and material intensive.
I'm only hoping that what I have is 2-octanol. Its smell is encouraging, ie, it's more oily and less pungent than the 2-octanone.
I wasn't sure I could even make 2-octanol by the OrgSyn procedure at such a small scale as 0.0055.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
The Volatile Chemist
International Hazard
Posts: 1981
Registered: 22-3-2014
Location: 'Stil' in the lab...
Member Is Offline
Mood: Copious
|
|
Be sure to do a second writeup when you're done; I've had a hard time following what actually works, and what is produced, but this certainly looks
like an interesting and doable reaction.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
This is a continuation of the above described synthesis of 2-octanol using the OrgSyn (B) procedure at 0.0055 scale.
After 5 batches the combined putative 2-octanol amounted to about 48 ml. This was dried over K2CO3 overnight. Today it was fractionally distilled.
A 100ml pot with electric heating mantle and a Vigreux column was used for the distillation. The pot and column were fully insulated with Al foil and
fiberglass blankets. No water was used in the condenser; it was set up as a contingency.

distillation of 2-octanol
Because of the high boiling point (lit: 178.5°C) a setting of 90% was required on the mantle. The liquid boiled evenly and steadily with no foam or
bumping. I took the heat up gradually and was concerned that I would generate tar when I found that a setting of 90% was required.
When sufficient heat was present the 2-octanol came over steadily at about 2d/s at a distillate temperature of 174-178.5°C per the literature range.
Most came over at 177-178°C.

2-octanol distillate temperature
The product was perfectly clear, as shown below:

2-octanol in receiver
The pot residue was only slightly discolored:

2-octanol pot residue
qualitative test results
1. A BB sized piece of sodium was placed in a ml of the 2-octanol. Profuse generation of H2 bubbles resulted.
2. Some acetyl chloride was added to a ml of 2-octanol. The smell of an ester was produced, ie, octyl acetate.
Both of the above tests are positive for an alcohol.
Discussion
Although my yield for the OrgSyn (B) procedure at my tiny scale was poor the product is of high quality. I now doubt that I would even have had to
distill it let alone fractionally distill it. I had given some thought to vacuum distilling (bp 86°C @ 20mmHg) due to concern over product
deterioration at the high bp. I decided against this for several reasons: (1) bumping might have been a real problem, (2) it's hard to keep the
pressure right where you want it, so the bp is lost as an indication of quality, (3) it's risky from a safety standpoint, and (4) it's more trouble to
set up.
Questions, comments, and suggestions are welcomed.
[Edited on 2-12-2015 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
The Volatile Chemist
International Hazard
Posts: 1981
Registered: 22-3-2014
Location: 'Stil' in the lab...
Member Is Offline
Mood: Copious
|
|
You test the alcohol for aldehydes and carboxylic acids and the like? Just to see if any impurities existed.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
No. The boiling point, color, and smell are all indicative of good purity. Even Sigma-Aldrich only offers 99% purity.
It would be nice to be able to check the refractive index but I don't have the instrument.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
The Volatile Chemist
International Hazard
Posts: 1981
Registered: 22-3-2014
Location: 'Stil' in the lab...
Member Is Offline
Mood: Copious
|
|
Great. Nice work.
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Arggh, just the usual Magpie quality work - which makes me envious more and more. (I'm still in the "gathering equipment and reagents" stage.) :-)
Good to know that even without vacuum you got a good looking (smelling?) product and little discoloration in the boiler flask.
I understand that you don't have (yet) a GC or NMR in your home but maybe a quick TLC run would also confirm the purity of the product. (Can anyone
reasonably expect more than one spot on TLC if the product is say 97%+ pure? Depends on the impurities I know, but would anyone expect anything in
this particular case?)
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
2-octylhydrogenphthalate
2-octylhydrogenphthalate was made using phthalic anhydride (PAN) and some of the 2-octanol synthesized above.
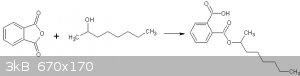
Reaction of 2-octanol with phthalic anhydride
I found 2 procedures for this synthesis, ie, that in Vogel, and that in OrgSyn (ref 1). I chose the OrgSyn procedure as it had the easiest workup.
The only potential downside to the OrgSyn procedure is the required reaction time of 12-15hrs. However, by using a PID controller to keep an oil bath
at a constant 110°C this required no attention.
I had several failures before discovering that my PAN was bad. Most likely it was instead phthalic acid (PA). So I painstakingly made 6g of PAN wool
by gathering it as it sublimed from PA heated to 185°C. I have posted a warning concerning this in Prepublication following my procedure for
converting PA to PAN. PA can be easily distinguished from PAN using chloroform (see Brewster, p. 119, forum library).
Procedure
5.3g (0.0405 mole) of 2-octanol was placed in a 25ml RBF. To this was added 6.0g of PAN wool (0.0405 mole). PAN wool is voluminous and had to be
stuffed into the flask as shown in the picture below:

25ml RBF with 2-octanol and phthalic anhydride
The RBF was closed with a glass stopper and placed in the silicone oil bath on a magnetic stirrer-hotplate. The oil was stirred magnetically; the oil
temperature was maintained at 110°C using the PID controller and a TC.
After about 3 hours most of the PAN had dissolved/reacted as shown in the picture below:

phthalic anhydride remaining after 3hrs
After 15hrs the product was poured into 342ml of water containing 6.1g of anhydrous Na2CO3. This formed the sodium salt. OrgSyn stated that if the
salt solution is hazy it is because of 2-octanone contamination. As you can see it is perfectly clear:

Na 2-octylphthalate solution
About 20ml of 6M HCl was then added to make the solution distinctly acid (est. pH 3-4). This formed the 2-octylhydrogenphthalate. At first a fair
amount of oil formed, floating on the surface. I left the lab to research what I might do to crystallize the oil. I could find no help. However, by
the time I got back to the lab about 1/2hr later the oil had disappeared turning into floating white solids, as shown below:

2-octylhydrogenphthalate
The solids were caught on a 7cm Buchner funnel, ground in a mortar with water, sucked dry, washed with water, sucked dry again, then placed in a
desiccator to dry.
The yield of crude product was near quantitative. Its melting point was found to be 46-47°C, even after washes with 95% ethanol and ether (OrgSyn
value: 55°C). I chose not to recrystalize it from glacial acetic acid or petroleum ether. OrgSyn recommended this if highly pure material is
required.
reference
1. Organic Syntheses, "D,L-s-octylhydrogenphthalate"
Comments, questions, and recommendations are welcomed.
[Edited on 11-12-2015 by Magpie]
[Edited on 11-12-2015 by Magpie]
[Edited on 12-12-2015 by Magpie]
[Edited on 12-12-2015 by Magpie]
[Edited on 12-12-2015 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
The OrgSyn reference in the above post should be:
"d- and l-OCTANOL-2"
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
The Volatile Chemist
International Hazard
Posts: 1981
Registered: 22-3-2014
Location: 'Stil' in the lab...
Member Is Offline
Mood: Copious
|
|
Nice final product. T'would be a nice collectable. Are the melting points of phthalates very variable with minor impurities?
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
2-octanol is racemic. The reason I made the ester was to allow separation of the enantiomers. But first I have to react it with a compound that
yields enantiomers having different solubilities.
Contaminants lower the melting point. How much depends on the molality of the contaminants. If you want to know more about this research
colligative properties.
All these edits mean I am a little rusty on my nomenclature.
[Edited on 15-12-2015 by Magpie]
[Edited on 15-12-2015 by Magpie]
[Edited on 15-12-2015 by Magpie]
[Edited on 15-12-2015 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
A value of 1.4225 was obtained for the refractive index of my synthesized 2-octanol. The value found on Wiki is 1.426.
It could likely be purified by fractional distillation.
edit: "A typical laboratory refractometer can determine the refractive index of a sample to a precision of ± 0.0002. However, small amounts of
impurities can cause significant changes in the refractive index of a substance. Thus, unless you have rigorously purified your compound, a good rule
of thumb is that anything within ± 0.002 of the literature value is a satisfactory match."
source: hanson@ups.edu
I don't think I really have my refractometer adjusted optimally yet as it is showing some chromatic aberration (color dispersion) in the viewing
field. I just bought it off eBay with no operating manual and I'm just learning to use it. It is an Atago (Japan) analog Abbé type manufactured ca
1963.
[Edited on 5-1-2016 by Magpie]
[Edited on 5-1-2016 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
| Pages:
1
2 |









