| Pages:
1
2 |
Praxichys
International Hazard

Posts: 1063
Registered: 31-7-2013
Location: Detroit, Michigan, USA
Member Is Offline
Mood: Coprecipitated
|
|
It doesn't really matter if your starting reagents are dry since the esterification reaction produces water anyway. To ensure completion, running the
reaction with continuous removal of water (dean-stark trap or similar) will work well.
Try using excess salicylic acid. Reflux for a few hours (with water removal, if possible*), let cool, then drip saturated bicarbonate solution until
exactly pH neutral or very slightly basic to convert the excess salicylate to the sodium salt. This will make the salicylate very soluble in what
little water is in the mix, and will probably force the product to layer out. If not, saturate with a neutral salt like sodium chloride. If it still
fails to layer out, extract with toluene.
Dry the product (or the extracted mixture) over a neutral drying agent (a vacuum desiccator would be ideal since the target has a high boiling point)
and then vacuum distill.
That's how I'd do it. Of course, I'd be making like 100g of the stuff.
* Water removal drives the equilibrium and will greatly increase yield. Just be careful if you use sulfuric acid or you might sulfonate some things if
the water content gets low. TsOH as a catalyst may be a better choice in that case, if you can make it.
|
|
|
Firmware21
Harmless
Posts: 33
Registered: 22-1-2015
Member Is Offline
Mood: Ņ͈̣̭̺̈ͬ͊̔i̓̿͑ͯ̂ͪ҉̸̺̀t͉̣͕͙̟̪̅͐͂̏͌ͭ͗̑͝ṙ̶̛̙̥̝̻̟̓ͬ̾ͧ͒͘ͅa͒͊ͯ̾̑̏̌̓̕҉͚͚͓͔͙͚̥t͆̌͑ͩ̐͗
͖̻̲̪̲̙͘ͅe͙͕͙̙̤̤̫̒ͩͣ̅̊̍̉͒ͬ́͟ͅs͈̬̮̥̻͂ͥͨ̂ͮͨ͒́͠ !!!
|
|
I'll try that next time (I'm not going for the 100g though).
However, unless I can improvise a dean stark with my current glassware set and figure out how to use it properly, I don't think I will be able to get
a continuous water removal. 
Also, I tried to extract the mixture with some ethylacetate, but I don't know yet if it catched anything.
|
|
|
Praxichys
International Hazard
Posts: 1063
Registered: 31-7-2013
Location: Detroit, Michigan, USA
Member Is Offline
Mood: Coprecipitated
|
|
The good news is that if you can distill, you probably already have one.
The main problem with this setup is joint leakage at the small flask. Grease it well and use your stiffest clip.
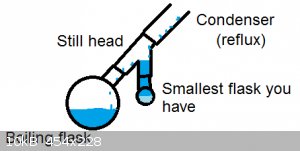
I sometimes use this setup when I know my dean-stark will need emptying every 10 minutes. You can trap a LOT of water with a setup like this. The
drawback is that it needs a lot of solvent, and you have to shut down to empty it. (Calculate water yield and select an appropriately-sized flask)
For your ester synthesis, you should run with NaCl dust or something in the trap to "salt out" the isopropanol that will distill into it with the
toluene, so the reactant gets returned to the flask but the heavier salt solution stays behind. You may be able to get away with just using
isopropanol as the solvent since it has a 91% azeotrope, and the salt will trap the water in the "dean-stark" and return only isopropanol.
Unfortunately you'd trade the use of toluene with having to fractionate the isopropanol from the final product, which is more difficult than removing
salicylate but not terrible since their boiling points are so far apart. Use a neutral, alcohol compatible salt in the trap like NaCl. If not, the
small amount of isopropyl salicylate that will come up with the toluene vapor (think: steam distillation) will be hydrolyzed if you use K2CO3 or
something, and trap salicylate while returning isopropanol, reducing efficiency.
If you decide to run with excess isopropanol (instead of salicylate), you will still need to neutralize the catalyst at the end, so the process for
extraction is essentially the same, just with far less base. There will also be an added step of fractionating off the alcohol either before or after
drying the mixture. I would recommend drying before fractionating since it may eliminate a lot of ternary/quaternary azeotropes that may form between
the components and water, since they're all polar to some degree. MgSO4 may work, but I would probably reach for the 3A or 4A sieves.
Sorry if this is too much detail. I get carried away sometimes. 
[Edited on 28-8-2015 by Praxichys]
|
|
|
Firmware21
Harmless
Posts: 33
Registered: 22-1-2015
Member Is Offline
Mood: Ņ͈̣̭̺̈ͬ͊̔i̓̿͑ͯ̂ͪ҉̸̺̀t͉̣͕͙̟̪̅͐͂̏͌ͭ͗̑͝ṙ̶̛̙̥̝̻̟̓ͬ̾ͧ͒͘ͅa͒͊ͯ̾̑̏̌̓̕҉͚͚͓͔͙͚̥t͆̌͑ͩ̐͗
͖̻̲̪̲̙͘ͅe͙͕͙̙̤̤̫̒ͩͣ̅̊̍̉͒ͬ́͟ͅs͈̬̮̥̻͂ͥͨ̂ͮͨ͒́͠ !!!
|
|
Thanks a lot ! I thought that I could distill the IPA as an azeotrope with water while adding anhydrous IPA with an addition funnel, but it should be
good with the improvised dean stark and sodium chloride, we'll see anyway. I hope a good old Vigreux column will be enough to separate the azeotrope
though.
Ps: IMO, there is no "excess details"
|
|
|
chemrox
International Hazard
Posts: 2961
Registered: 18-1-2007
Location: UTM
Member Is Offline
Mood: LaGrangian
|
|
phenethyl anthranillate smells like oil of musky rose. I would be an even cooler chart if the pictures were clickable.
"When you let the dumbasses vote you end up with populism followed by autocracy and getting back is a bitch." Plato (sort of)
|
|
|
chemrox
International Hazard
Posts: 2961
Registered: 18-1-2007
Location: UTM
Member Is Offline
Mood: LaGrangian
|
|
Praxichys I have a couple of Dean Starks with stopcocks. I'll sell one if you like. Say $20.
[Edited on 28-8-2015 by chemrox]
"When you let the dumbasses vote you end up with populism followed by autocracy and getting back is a bitch." Plato (sort of)
|
|
|
Praxichys
International Hazard
Posts: 1063
Registered: 31-7-2013
Location: Detroit, Michigan, USA
Member Is Offline
Mood: Coprecipitated
|
|
Mine has a stopcock, but it's a pain to keep draining 10ml of water every few minutes. I sometimes use the other type of setup because I can just get
it going and leave it for a few hours, and it's done when I get back. Drying wet, deliquescent items I need upwards of ~150ml water capacity
sometimes. It's nearly impossible to get the rate perfect and just leave the stopcock open slightly.
$20 is a good deal for one of those. Do you happen to have one with a capacity greater than 15ml? I'd take a 50 if they even make them that big.
|
|
|
Brain&Force
Hazard to Lanthanides
Posts: 1302
Registered: 13-11-2013
Location: UW-Madison
Member Is Offline
Mood: Incommensurately modulated
|
|
Quote: Originally posted by Etaoin Shrdlu  | | Also, propyl acetate. Pear or not pear? I know it's supposed to be pear. It's supposed to be quintessential pear. But it just smells horrid
to me, even in small quantities. Horrid in a way that makes me understand what people mean when they say they can't stand organic solvents.
|
I have some n-amyl acetate and I find that to be the quintessential, unwashed, raw pear scent, with slight hints of banana, apple, and pear. It's
dissolved in 1-pentanol, though, which might affect the scent enough to produce those other notes.
At the end of the day, simulating atoms doesn't beat working with the real things...
|
|
|
Firmware21
Harmless
Posts: 33
Registered: 22-1-2015
Member Is Offline
Mood: Ņ͈̣̭̺̈ͬ͊̔i̓̿͑ͯ̂ͪ҉̸̺̀t͉̣͕͙̟̪̅͐͂̏͌ͭ͗̑͝ṙ̶̛̙̥̝̻̟̓ͬ̾ͧ͒͘ͅa͒͊ͯ̾̑̏̌̓̕҉͚͚͓͔͙͚̥t͆̌͑ͩ̐͗
͖̻̲̪̲̙͘ͅe͙͕͙̙̤̤̫̒ͩͣ̅̊̍̉͒ͬ́͟ͅs͈̬̮̥̻͂ͥͨ̂ͮͨ͒́͠ !!!
|
|
Fellow chemists, i present you the magnificent derp-stark :p
I tested it on a citric acid/IPA esterification: the system works pretty well (no leaks or whatever) but I didn't get the expected separation (salting
out) in the trap. Maybe there was too much IPA for the amount of water generated.
I used about 20 g of citric acid (monohydrate) and an extreme overkill of 150 ml IPA, knowing at least 70 ml were required to fill the trap ( + 3 drop
of conc. H2SO4) The trap was half filled with table salt.
Refluxed it for 4 hours.
I then tried to neutralize it with NaHCO3, but it fizzed way too much for the amount of catalyst used. Either caused by unreacted acid and/or free
carboxyl group in the mono/di isopropyl citrate...

|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
NaCl will only salt water out of IPA up to about 90%. After that point, the salt just sits there and stronger dessicants are needed.
|
|
|
chemrox
International Hazard
Posts: 2961
Registered: 18-1-2007
Location: UTM
Member Is Offline
Mood: LaGrangian
|
|
Attempted synthesis of deschloroloperamide propanoate. This is involves a hindered OH group. I dissolved 14 g deschloroloperamide in 300 ml DCM and
refluxed at ~ 41*. I added 2.8 ml propionyl chloride and observed bubbling and a slight increase (~2*) in reflux temp. Continued reflux for another 3
hrs. Extracted with 1M NaOH and dried over NaSO4. Cryx in dish over night. Collected product and took mp.s; deschloroloperamde MP 102-104*C, product:
90-104 *C. I suspect partial esterification occurred but IR is down.
"When you let the dumbasses vote you end up with populism followed by autocracy and getting back is a bitch." Plato (sort of)
|
|
|
DraconicAcid
International Hazard
Posts: 4356
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Extracted with sodium hydroxide? Wouldn't that likely hydrolyze the ester?
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
chemrox
International Hazard
Posts: 2961
Registered: 18-1-2007
Location: UTM
Member Is Offline
Mood: LaGrangian
|
|
No not 1M as a wash. Corrections: mp was wrong.. to high temp and too much of a hurry
mp was 92-94 vs 102-104 for the starting material. Separation by TLC was ok but mixed with the some of starting spot. Polar solvent mixture- the ester
migrated more slowly-seems consistent with mp however the mixture is troubling so how to proceed? More acyl halide? longer time? catalyst? which one?
[Edited on 9-6-2016 by chemrox]
"When you let the dumbasses vote you end up with populism followed by autocracy and getting back is a bitch." Plato (sort of)
|
|
|
| Pages:
1
2 |










