| Pages:
1
2
3
4
5 |
Cheapskate
Harmless

Posts: 27
Registered: 3-2-2005
Member Is Offline
Mood: Just fine, than
|
|
Holy Cow, using an expresso machine never occurred to me. Right now I have the large volume of water extraction running, but the old expresso machine
will come out for the next try.
Talk about high pressure steam extraction! Thank you very much, I'll report back on both of the current ideas.
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
| Quote: | Originally posted by Eclectic
Side reactions due to everything other than nicotine that is in the tobacco.
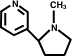
You don't think the pyrrolidene ring is subject to attack? I'm thinking that some of those carbons could give up H+ .
|
N-methyl-pyrrolidine has no acidic hydrogens. If there was a hydrogen instead of the methyl group on the nitrogen, then it would have been another
story, but it would still take BuLi or simillar to deprotonate it.
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Pull hydrogen from the attachment point to the pyridine ring: nitrogen ylide/imine, resonance stabilized with pyridine ring.
I think that pyrrolidene ring is going to fall right apart if you look at it crosseyed under strongly alkaline conditions.
http://clem.mscd.edu/~wiederm/401chp/unit3chp/ylideschp/page...
We already know that nicotine is very easily oxidized under pH neutral conditions.
[Edited on 20-2-2005 by Eclectic]
[Edited on 20-2-2005 by Eclectic]
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
| Quote: | Originally posted by Eclectic
Pull hydrogen from the attachment point to the pyridine ring: nitrogen ylide/imine, resonance stabilized with pyridine ring. |
Huh?
| Quote: | I think that pyrrolidene ring is going to fall right apart if you look at it crosseyed under strongly alkaline conditions.
http://clem.mscd.edu/~wiederm/401chp/unit3chp/ylideschp/page...
We already know that nicotine is very easily oxidized under pH neutral conditions.
[Edited on 20-2-2005 by Eclectic]
[Edited on 20-2-2005 by Eclectic] |
In the link that you provided there is a positive charge on the nitrogen, this is the reason why the base react as it does, since the resulting
negative charge can be stabilized (lone pair forms on the nitrogen). Do you see a +1 charge on any nitrogen in nicotine?
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
The imine should form with no problem.
You aren't going to be happy without experimental verification, are you?
Reflux some nicotine with NaOH, assorted carbohydrates and other biologicals while exposing to air and see how much of the nicotine you can
recover.
I might be wrong, it's been many decades since my chem courses, ...but I don't think so.
[Edited on 20-2-2005 by Eclectic]
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
| Quote: | Originally posted by Eclectic
The imine should form with no problem. |
No, the imine will not form.
| Quote: | | You aren't going to be happy without experimental verification, are you? |
Anything that makes sence would have been fun. I just find it hard to belive that a hydrogen attached to a carbon (pKa around 50) can be stripped off
using OH (pKb ~16)... The whole discussion is getting really boring and repetative...
| Quote: | | Reflux some nicotine with NaOH, assorted carbohydrates and other biologicals while exposing to air and see how much of the nicotine you can
recover. |
I'm not intersted in nicotine, so I won't do that...
| Quote: | | I might be wrong, it's been many decades since my chem courses, ...but I don't think so. |
Okidoki..
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
"I just find it hard to belive that a hydrogen attached to a carbon (pKa around 50) can be stripped off using OH (pKb ~16)..."
And yet that is exactly what happens in aldol and claisen condensations.
So is the number 2 carbon in the pyrrolidene ring more or less acidic than average? I was thinking more acidic, but I might have it backwards.
Methyl iodide methylates nicotine on the pyridine nitrogen if that gives any clue.
http://www.medicine.uiowa.edu/frrb/education/FreeRadicalSp01...
(page 5)
[Edited on 20-2-2005 by Eclectic]
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
| Quote: | Originally posted by Eclectic
And yet that is exactly what happens in aldol and claisen condensations. |
In such reactions this is possible since you have a carbonyl group alpha to the carbon atom bearing the acidic hydrogens (that's the reason
they're acidic). When you deprotonate the alpha carbon, the negative charge can be stabilized, ending up on the oxygen and you get an enol.
| Quote: | | So is the number 2 carbon in the pyrrolidene ring more or less acidic than average? |
Since you have an electon donationg group (an amine with a lone pair of electrons) this would lead to even further destabilization of the negative
charge that might result from deprotonation of the adjecent carbon. For instance the base streangth of BuLi goes: tert-BuLi > sec-BuLi >
n-BuLi.. Reason for this is that methyl groups are electron donating, this destabilize the negative charge on carbon and the tertiary base is stronger
(the more electron-donating groups around the carbanion - the less stable it is). This is opposite for carbocations (acids), electron-donating groups
stabilize the positive charge, hence acids and bases are eachothers opposites..
| Quote: | | Methyl iodide methylates nicotine on the pyridine nitrogen if that gives any clue. |
Yes, and that product would be reactive toward nucleophiles as it would contain C=N-Me (+), this would make it electrophilic as Nu: (-) ---> C=N-Me
(+) gives Nu-C-N-Me.. The positive charge makes a lot of difference as it is powerfully electron-withrawing, comparable to a nitro group...
But admittedly there is a reaction in Vogel's third ED where pyridine itself reacts with NaNH2 (very strong base), so it is possible but NaOH is
not powerful enough...
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
I yield. I mistakenly thought the amine and pyridine ring were electron withdrawing, and the lone pair on the nitrogen would stabilize the imine.
|
|
|
Cheapskate
Harmless
Posts: 27
Registered: 3-2-2005
Member Is Offline
Mood: Just fine, than
|
|
OK, so it doesn't appear that I ruined it by being over zealous with the NaOH.
My experiments with a LOT of water are proceeding, but I suspect I just wasted a bunch of materials. There is a bad problem with emulsions. Looks like
the alkaline nature of the raw material and the fats don't mix well. I get a soapy mess if I mix a non-polar with the water extraction.
Now, I've seen emulsions before, but this one is terrible. If you wait long enough, it solidifies. Not quite into a wafer, but currently I have a
two inch emulsion in my sep funnel that resists gentle shaking.
So, not only does one have to deal with the swelling tobacco that just won't filter worth a damn, you get the emulsion from hell. No, adding more
solvent doesn't help, nor does adding more water. No wonder all the schoolroom extractions start with something besides water.
I tried the idea of an expresso machine. I loaded 20grams of tobacco in it with 4 cups of water and let it go. I got about two cups out before it
plugged the filter in the expresso machine. I vented the machine and pulled the coffee holder off and broke up the tobacco cake and fed another two
cups of water through the cake. I now have an extremely dark 500ml of water and tobacco extract. Interesting smell, but not much different from a
normal water extraction. This is just setting on a shelf waiting for me to do something with it.
However while I was setting there staring at it I had an idea. I took a few grams of tobacco and put it in a tube with maybe 10 ml of acetone. I
heated this to boiling after adding a chunk of NaOH. The idea is to extract what I can out with the acetone. I expected the acetone to turn black like
the water since it's polar as well, but it only took on a yellowish color.
This, when evaporated, left a yellow tar. I dissolved as much as I could of the tar into 10 ml of water. This left an insoluble resin like substance
and gave me some slightly yellow water. Now I know there isn't enough nicotine in this water to actually do anything with, but I was flat amazed
that the acetone didn't pick up more from the tobacco.
Could this be a clue? Dry some acetone, use more tobacco and see if I've found a way to leave most of the undesirables behind? My experience has
been that nicotine is extremely solubile in acetone so I should have gotten it out of the small sample of tobacco.
I suspect I'll still acidify the bunch of water extract I have and reduce the volume to see what's in it, but what a mess that will be.
More thoughts, ideas? Keep em coming, I'm listening.
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
Heating acetone with NaOH and nothing else save traces of water will give you colored condensation products, though I don't know what
that'll yield on evaporation. I would run a comparison with acetone and a bit of NaOH, sans tobacco, to get an idea of how much gunk you're
introducing this way.
Swelling and emulsions seem to be major problems, and swelling will (presumably) not be triggered by nonpolar solvents. Soxhlet extraction with a low
BP nonpolar? Grinding it to dust, then heating the tar (ha ha) out of it in a highish-BP hydrocarbon like toluene or xylene? Maybe ball-milling it
with dry NaOH before mixing and heating with hydrocarbons?
Or here's an odd idea: use p-dichlorobenzene (mothballs) as your hot extraction solvent. It has a highish 174 BP but sublimes readily, so can be
separated from your extract more easily than xylene or toluene. I'm going to make the unsubstantiated guess that, all else being equal, higher
solvent temperatures will give you better extraction of the plant matter. I imagine the real trouble would be keeping it hot while filtering, and not
being choked by all those mothball fumes when working with it hot and in the open.
Hmm, make some sort of "teabag" to hold the tobacco? Tobacco and mothballs go in sealed jar, sealed jar goes in sandbath, tobacco is
extracted by molten p-dichlorobenzene, jar is afterward allowed to cool to room temperature, jar is smashed and solid p-DCB mechanically separated
from the teabag, p-DCB is sublimed away with moderate heat or maybe vacuum. For that matter naphthalene could probably do the same trick, depending on
what sort of mothballs you find.
Or if you don't like high temperatures, what about ball-milling it with your solvent for an extended period of time?
What about dry distillation in inert gas? It's probably not something you'd like to use good glassware for. Maybe actually
dissolve/dry-distill the tobacco in NaOH/KOH eutectic? I'm sure it will take apart the plant material like nobody's business, but it's
a powerful oxidizing environment if you can't exclude air (even then I'm not sure).
Dry distillation variation: mix dry, clean sand, table salt, or anhydrous MgSO4 with powdered tobacco, place in a tube or jar, purge jar with inert
gas, and heat. What can you (nonpolar) solvent extract from the mixture of sand and cooked tobacco afterward? With a tube, can you stick the tobacco
at the bottom, dry powder over it, and heat only at the bottom so that products are trapped in the progressively-cooler solids above the heated
region? Do you pour solvent over the whole mess and heat it again after the really high-temperature phase? I'm imagining ways to trap volatile
products of dry distillation without actually using good glassware or custom apparatus.
Maybe get a secondhand centrifuge and use it to overcome the difficulties with aqueous solutions?
Are there any microorganisms that would attack the leaf matter yet leave the nicotine intact, so that there's nothing left to swell and clog
filtering attempts?
Well, that's my grab-bag of wild ideas for the night.
PGP Key and corresponding e-mail address
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Well yes, you probably didn't destroy the nicotine with NaOH, but the idea of making all kinds of crud with everthing else in the tobacco is
still valid. pH9-11 is plenty basic to free up most of the nicotine.
With the expresso machine run, did you acidify the water with anything? Maybe try white vinegar as extractant. 1-2 cups will probably extract the
bulk of the nicotine. Your initial water extract needs to be acidic to extract soluble nicotine salts. If you want to steam distill the free
nicotine, you would make the water alkaline, 1-3% Na2CO3 would have a pH 11-11.5. I think there would be serious risk of poisoning from the vapors if
you tried that with the expresso machine.
(If you boil baking soda and water you end up with a sodium carbonate solution)
[Edited on 22-2-2005 by Eclectic]
|
|
|
sparkgap
International Hazard
Posts: 1234
Registered: 16-1-2005
Location: not where you think
Member Is Offline
Mood: chaotropic
|
|
Eclectic,
QUOTE:
...I think there would be serious risk of poisoning from the vapors if you tried that with the expresso machine...
Now that would be a novel method of nicotine administration. 
Polverone:
Use p-dichlorobenzene? Seeing that he intends to consume the nicotine thus extracted, would it really be worth it to attempt extraction with a suspect carcinogen? Now naphthalene, hmm...
Cheapskate:
Just to confirm, do you do these extractions with fresh tobacco leaves or dried tobacco leaves?
sparky (^_^)
"What's UTFSE? I keep hearing about it, but I can't be arsed to search for the answer..."
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
If you cant filter it, then you shouldn't grind so finely, or you should change your filter to something like mesh or cloth. I had no problems in
my experience making my own insecticide years ago.
I'm not so sure that adding a strong base to plant material is a good idea.
The extract you have should already be acidic, although Merck says that nicotine is soluble in water below 60C, I'd still add a little acid as
suggested before.
[Edited on 22-2-2005 by S.C. Wack]
|
|
|
Cheapskate
Harmless
Posts: 27
Registered: 3-2-2005
Member Is Offline
Mood: Just fine, than
|
|
Every attempt has been done with dried tobacco products. I'm currently working my way through a bag of tobacco dust that was designed as an
insecticide. I don't have access to fresh tobacco leaves.
Even though they're dried and powdered, they have a water content. My attempts at dry distillation using a cold finger turned up a lot of water.
Mothballs are a compelling idea because they are so out-of-the-box, but I suspect they would be the same as using a low temperature solvent such as
ether and just letting it evaporate off. There is the advantage of being able to heat it, but wouldn't that be the same as an ether reflux?
Actually, I started to try the ether idea, but chickened out due to the flamability problem. DCM would be a possibility since it's non polar and
has a low boiling point, but I have to distill that stuff out of paint remover to get it. A painful, long, tedious process that I wanted to avoid if
possible. The school book extractions often start with methanol as the first extraction, but my experience with alcohol (95%) was that I had a swollen
ball of tobacco and couldn't get the alcohol loose. Now that may have been the 5% water, but I suspect it worked like water and caused swelling.
I haven't actually tried methanol yet since I don't know what it its solubility is with other solvents. It could be a problem separating
from something else. I guess I could just evaporate it away.
I used to have a link to a solubility chart of various solvents in other solvents. It got lost somewhere. Does anyone have such a thing?
|
|
|
unionised
International Hazard
Posts: 5126
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
Well, everyone else has had a go so here's my 2c worth.
The fats are a pain in the neck if you do an alkaline extraction, they make soaps and the emusions never settle.
A couple of thoughts, 1 use calcium hydroxide and make insoluble soaps that are much less effective emulsifiers. or
2 Don't do an extraction. I read that the commercial process uses steam distillation of an alkaline extract.
Not many of the compounds present in plants will steam distil.
If you take a flask, put the tobacco in it and wet it with slightly alkaline (you don't want to get things too alkaline, god knows what other
reactions you might get) water saturated with salt, then distill this you should get steam, at a higher temperature than normal because of the salt,
(and, because of the salt, the soaps won't dissolve so well so it will foam less). This should carry over the nicotine (You may need to add a
dropping funnel to the aparatus so you can add more water to get all the product over.)
That should give a crude distillate with the prduct in water. Acidify it (again, there's no point in overdoing it) and boil it down. That should
remove a lot of the other organic trash that is not an alkaloid, but is steam volatile. Re basify it and re distill over the nicotine (and other
steam-volatile alkaloids).
If all the assumptions I have made all work you should end up with crude nicotine in water. Add HCl and leave it somewhere warm for the water and
excess HCl to evaporate, you should end up with nicotine hydrochloride.
It won't be very clean, but it should do the job.
|
|
|
JohnWW
International Hazard
Posts: 2849
Registered: 27-7-2004
Location: New Zealand
Member Is Offline
Mood: No Mood
|
|
Besides nicotine, tobacco also contains other alkaloids similar to nicotine, for example, isomers in which the tetrahydropyrrole ring is attached at
the 2- or 4- position to the pyridine ring and/or via the 2-position on the tetrahydropyrrole, and/or the N-CH3 group is replaced by --NH or N-CH2CH3
or others, and/or there are substitutents on the pyridine or tetrahydropyrrole rings. Besides nicotine, nornicotine ("nor" meaning without
the -CH3 group), anatabine and anabasine are the principal minor alkaloids present. Another is nicotyrine. They are all chiral, having optically
active enantiomers, which may react differently with some reagents. Although the raw tobacco alkaloids are 98% nicotine, the minor ones would have to
be somehow taken into account, or removed e.g. by chromatography, if one wanted to use raw nicotine extracted from tobacco as a reagent for syntheses.
See http://www.tobaccopedia.info/leaf_chemistry/alkaloid_biosynt... , http://www.tobaccopedia.info/leaf_chemistry/the_influence_of... , http://www.ash.org.uk/html/regulation/html/additives.html , http://www.trdrp.org/research/PageGrant.asp?grant_id=2142 , http://www.sensir.com/newsensir/AppNotes/app030.pdf , http://pages.towson.edu/jsaunder/Saunders%20Publications/15.... , http://www.shaman-australis.com/~auxin/tobacco.html , http://www.ndp.govt.nz/tobacco/cigaretteaddictiveness.PDF , http://www.ndp.govt.nz/tobacco/cigaretteaddictiveness_final%... , http://www.ash.org.nz/pdf/Smoking/Tobacco/Nicotine.pdf , http://www.foodstandards.gov.au/_srcfiles/P278_Nicotine_&... , http://www.cdc.gov/tobacco/sgr/sgr_1988/1988SGR-Chapter%202.... ,
http://doi.wiley.com/10.1002/(SICI)1520-636X(1999)11:1%3C82::AID-CHIR14%3E3.0.CO;2-C ,
http://doi.wiley.com/10.1002/(SICI)1520-636X(1996)8:4%3C295::AID-CHIR1%3E3.0.CO;2-F , http://tobaccodocuments.org/product_design/2022157406-7416.h... , http://arjournals.annualreviews.org/doi/pdf/10.1146/annurev.... , http://www.hc-sc.gc.ca/hecs-sesc/tobacco/pdf/T-301e4.PDF (Determination Of Alkaloids in Tobacco) , http://www.isc-newsletter.com/newsletter/nov04/lin/art3.pdf (Determination Of Alkaloids in Tobacco by Extraction).
Nicotine alkaloids are also found in the leaves of the coca plant, see http://www.answers.com/topic/nicotine .
They become carcinogenic through the formation of nitrosamines, e.g. N-nitrosonornicotine , http://www.smoke-free.ca/factsheets/pdf/TSNAfactsheet.PDF .
[Edited on 22-2-2005 by JohnWW]
|
|
|
wallbanger
Harmless
Posts: 9
Registered: 22-2-2005
Member Is Offline
Mood: wallbangical
|
|
ethanol alkaloid extraction
Hey guys,
This has already been mildly discussed, but I think is the simplist way.
Matierials:
1.75 liters of greater than 150 proof ethanol (everclear, moonshine)
2 mason jars
1 mason jar lid
a non-gas stove
cheesecloth or old T-shirt
3 or 4 packs of cigarettes
Procedure:
Start by butchering your lovely cancer inducing cigarettes by creating an incision at the top and work your way down to the filter. Extract and
chop the resulting tobacco with a knife untill it is very fine. Put all of this tobacco into the mason jar and pour in you ethanol solution of choice.
Don't fill the jar, just put enough in so the tobacco particles float around freely.(Sorry for lack of definitive measurments) Let this solution
sit for approx. 30-60 minutes. The longer this is done the better. I do three myself.
After you feel the solution has sat long enough, place the cheesecloth or T-shirt over the other mason jar. Pour the contents of jar one into jar
two through your filter. Pick up your filter and SQUEEZE all the liquid you can from the tobacco pulp. Repeat the process as much as you want.
Once your second mason jar has been filled to the amount of your liking, pour it into a wide bottomed pot or pan and place over a non-gas stove at
a low to medium-low heat. Ok, I can't stress this enough, DO NOT USE A GAS STOVE!!! The ethanol vapors WILL combust in your FACE! Agitate the
solution with a utensil of choice for 1.5 hours or untill the solution is thick and doesn't smell of alcohol. If you have gotten this far and
didn't screw up, congratulations! You now have around 72% pure tobacco alkaloid extract and according to JohnWW, 98% of the alkaloids are
nicotine. So your final product should be approx. 66.24% pure.
Not bad for a bad procedure for higschool freshman eh?
\"But did you, in your three-piece psychology and 1950\'s techno brain, ever take a look behind the eyes of the hacker?
Did you ever wonder what made him tick,
what forces shaped him, what may have molded him?
I am a hacker, enter my world...\"
(\"The Conscience of a Hacker\", The Mentor)
|
|
|
Cheapskate
Harmless
Posts: 27
Registered: 3-2-2005
Member Is Offline
Mood: Just fine, than
|
|
I, sort of, tried this. What happened to me was: I used everclear 95%, not the 73% stuff in California and extracted about 50 grams of tobacco three
times with it. I didn't squeeze it until the last extraction. I just let it drain through as best it could.
The idea was that I didn't want to lose much of the chopped up tobacco to the filtering cloth. That may have been a mistake since it took forever
to get the alcohol to drain out. I did this extraction three times. I filtered the combined solutions three or four times to remove as much of the
solids as possible.
Now I had a dark solution of alcohol that I evaporated down slowly to a syrup and raised the temperature on the syrup to 100C to be sure all the
alcohol was evaporated away. This I then acidified to make nicotine.hcl and washed with ether to remove fats, oils and such.
I was in the middle of emulsion hell. Notice that there wasn't any base addition to create soaps, it just came on its own. I separated the ether
as best I could and evaporated it to see what I got. There wasn't anything in the ether worth bothering with. I then raised the temp on the
original extraction back to 100C to get rid of the ether.
I based it and extracted with toluene. I did get nicotine, but it was heavily laced with tars and other stuff. The heavily poluted nicotine was put
in a tube where I was collecting the results of my various experiments and dissolved in acetone.
After a number of relative failures using various techniques I did an acid base extraction on the combined results and got a few grams of nicotine
that is still somewhat impure, but not at all bad.
What I'm searching for (the holy grail) is a method that avoids the darn emulsions, gets at least half the available nicotine and doesn't
take a week per session. Part of this has to be treating the extraction with a strong base to free the nicotine from the various salts formed by
nature.
I'm going to go back to the alcohol method eventually and see if I can avoid the emulsion by taking the remains up in a non-polar solution after
drying to a tar. I suspect it will leave the nicotine salts behind though.
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
You don't need a really strong base. Sodium carbonate is good enough.
Use acidic water (vinegar) expreso machine extraction, make alkaline with sodium carbonate, distill off 1/2 the amount of your initial aqueous
extract, extract the distillate with nonpolar solvent, maybe brake cleaner (perchloroethene). Nicotine is in the perc. layer (bottom).
Hydrodistillation leaves all the organic crud from the initial aqueous extraction behind.
Also, if you are still using the tobacco insecticide dust, it may have soaps and other things added to it to help it stick to damp plants. Start with
cigarette or pipe tobacco. If you never plan to kick the nicotine habit, you might try growing your own Nicotina rustica. It's supposed to be
MUCH more potent than commercial tobacco.
[Edited on 23-2-2005 by Eclectic]
|
|
|
Cheapskate
Harmless
Posts: 27
Registered: 3-2-2005
Member Is Offline
Mood: Just fine, than
|
|
One thing I'm going to start a separate thread on in the organic chemistry forum is micro distillation. At some point in this effort I will
succeed and actually have some of this stuff. It would be good to distill it, but there won't be a half liter to mess with, I'll be looking
at distilling, say 10ml.
Now I know how to do this if the lower boiling fraction is the part I want, but what if I want the higher fraction? I know that I'll have to use
vacuum, but what kind of set up will I need. My 24/40 set up is just too darn big.
You don't need to answer here, I'll be putting it over there and I don't want to mess folks up by double posting.
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Anyone have the vapor pressure of nicotine at 100C? I have 17mm-hg at 125C. I'm guessing at least 7.6 mm at 100C, so 100ml water as steam
should carry over ~1 gram nicotine. Probably at most 2% nicotine in commercial tobacco, so 50g tobacco would have about 1 gram.
|
|
|
unionised
International Hazard
Posts: 5126
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
I have a couple of refs that gives 1mm at 68C and 12 mm at 116C
To a fair approximation, the log of the pressure is proportional to the absolute temperature. That should be good enough to interpolate.
|
|
|
fritz
Harmless
Posts: 49
Registered: 29-11-2003
Member Is Offline
Mood: No Mood
|
|
I researched some old sources and found the following :
Nicotine is found especially in the leaves and seeds of the various tobaccos (0.68-8%). There it is bond to malic acid.
EXTRACTION:
Tobacco leaves are mixed with water for one day. The mixture is heated to 160-180 °C (by introduction of steam or over free flame) The warm mass is
squeezed out and the mixture is treated again as above. The extracts are then evaporated to 1/3 of their original volume. CaO (10% of the leaves´
weight) is added and the mixture is steam-distilled. The end of the distillation is reached when the distillate lost its characteristic smell of
nicotine (this is a better way then checking with indicators). The distillate is acidified with a measured amount of oxalic acid and evaporated to
dryness. The residue is treated with an equivalent (referring to the acid) amount of KOH (as concentrated solution). The crude nicotine separates as
an oily layer on top. The oil is separated and the aqueous phase is extracted with Et2O. Both Fractions are united and distilled under H2. The
destillate is again acidified with oxalic acid and the salt is washed with Et2O, treated with KOH and distilled (again under H2). To remove most of
the impurities (mainly. Et2O and NH3) the mixture is heated slowly for 2-3 °h to 210 °C. The fraction from 230-250 °C is again distilled and pure
nicotine should come over at 240-242°C.
QUALITATIVE REACTIONS:
solution in CHCl3 gives a precipitate with oxalic acid wich is soluble in EtOH (ammonium oxalate is not)
HgCl2 gives a white precipitate which is soluble in HCl
I2/KI gives a yellow precipitate
Picric acid gives a yellow precipitate (m.p. 218 °C) soluble in HCl
[translated from: Einführung in das Studium der Alkaloide, Dr. Icilio Guaresci; 1896]
PURIFICATION OF (CRUDE) NICOTINE:
Crude nicotine is dissolved in an excess of H2SO4. The solution is shaken with Et2O and the nicotine is freed by KOH.
QUANTITATIVE DETERMINATION (in tobacco):
Tobacco is dried for 1-2 h at 50-60 °C. 20 g of it are ground with 10l NaOH-solution (6 g NaOH, 40 ml water and 60 ml EtOH 95%). The mixture is
extracted in a ´TOLLEN´s appartus´ (never heared of this!) with Et2O. The the ether is carefully (but not completely) evaporated. The residue is
mixed with 50 ml 0.1 n NaOH and steam distilled until only 25 ml remains in the flask. 100 ml of the distillate are titrated with H2SO4.
[translated from Beilstein]
SOME SOLUBILITIES:
water EtOH Et2O other
nicotine ∞ ∞ ∞ soluble in:
n-pentanol; CHCl3; petrol ether
-hydrochloride soluble soluble soluble
-salicylate
(m.p. 117.5 °C) soluble soluble soluble
-tartrate
(m.p. 88-90 °C) soluble soluble soluble
Beilstein gave the same method for extraction as the first source. The first source also gave some other slightly different methods (H2SO4 instead of
oxalic acid, precipitation from Et2O-solution with oxalic acid). Also there are mentioned some other qualitative reactions but these are the typical
methods of precipitating various alkaloids (PtCl2; AuCl3, etc.)
An interesting discovery is that tobaccos of poor quality should have the highest nicotine content and reverse.
To Cheapskate:
You mentioned difficulties with water/tobacco-mixtures. I didn´t make this experience when I extracted tobacco (nearly dry, from cigarettes) with
some acidified water. I got something similar to tea. Otherwise if you are going to distil the extract its consistency doesn’t matter.
Very at the beginning you mentioned problems with getting solvents off the nicotine. In general this should not be a serious problem. If you have
access to liquid nitrogen and some kind of vacuum-pump you may try to ´degas´ the nicotine: take the flask with the nicotine (should be round-bottom
with ground joint) and connect to the vacuum pump. After reaching minimal pressure carefully cool with liquid nitrogen until the whole mass
solidifies. Take the flask out of the nitrogen and wait (still the flask under vacuum) until its content is liquefied again and no more gas is
bubbling out of the liquid. Repeating this a few times you should get rid of nearly every impurities (with lower b.p. than nicotine). Another method
(perhaps more suitable for amateur-labs) is again to apply vacuum to the flask and then carefully heating the contents. Nicotine with a b.p. of about
250 °C will boil at 75 °C at a pressure of 1.33 mbar/1 mmHg. With a common water jet pump you should reach between 13 and 26 mbar (10-20 mmHg) and
the nicotine boils between 115-140 °C in this range of pressure. Again: repeating this step a few times (or one long-time drying) will remove all
solvents (especially the hydrocarbons you used). This method (as well as the above) are also suitable for handling small amounts.
For controlling purity (as well as for purification itself) I would suggest using chromatography. If you have the equipment and some experience
column-chromatography should be your first choice for purification!
Here some Rf-values:
mobile phase (v/v) Rf
EtOAc/MeOH/conc. NH3 (85/10/5) 0.61
MeOH/conc. NH3 (100/1.5) 0.54
CHCl3/MeOH (9/1) 0.43-0.45
MeOH 0.39
acetone 0.13
n-hexane/acetone (8/2) 0.04
Toluene/acetone (95/5) 0.01
adsorbent in all cases is silica
One great disadvantage is: you will need LOTS of solvents/! But this would need only one perhaps expensive investment as you may redistill (and reuse)
all solvents.
For controlling purity (or checking the fractions during column-chromatography) TLC may be useful. Detection of the nicotine may be carried out by
viewing the plate under UV-light or by spraying with ´DRAGENDOFF-MUNIER-reagent´. This consists of two stock-solutions:
A: bismuth-subnitrate: 17 g; tartaric acid: 200 g; water: 800 ml
B: KI: 160 g; water: 400 ml
Before using both solutions are mixed in the ratio 1:1 and 50 ml of the mixture are diluted with 100 g tartaric acid and 500 ml water. Nicotine should
be visible as red spots.
BUT: this is a common reagent for alkaloids! So it is important to check the Rf-values of the determined spots. These should be approximately as given
above.
[Edited on 24/2/2005 by fritz]
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Looks like my estimate is in the ballpark for the vapor pressure of nicotine at 100C then. I forgot to correct for molar weight though, so it may
take as little as 15ml water to steam distill 1 gram nicotine. 100ml should be more than enough to carry over all traces of nicotine. That makes
hydrodistillation look very atractive for the inital purification. No need to inject steam, just make the initial extract alkaline (ph 11 or higher)
and distill off 1/2 the water.
[Edited on 24-2-2005 by Eclectic]
|
|
|
| Pages:
1
2
3
4
5 |









