| Pages:
1
2 |
DJF90
International Hazard

Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Wikipedia gives the melting point as 50 *C. I suspect you're having trouble because your product is impure, or because you have residual toluene
(which is the same problem, actually).
Ideally you'd stick your sample on the high vac line to try and remove any residual toluene. A better (and more accessible) idea may be to concentrate
your product from methanol (methanol and toluene azeotrope...), and if you still have problems (possibly at that point from residual methanol) then
concentrate again, this time from DCM.
I have this same issue at work sometimes. Product is a residue on the walls of the vial after concentrating fractions (chromatrographic purification)
containing MeOH-EtOAc. A quick strip from DCM then leaves me with the familiar foamy solid I'm after.
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
Thanky DJF90, it is 2-Hydroxy-5-methoxybenzaldehyde not 2,5-Dimethoxybenzaldehyde which has its mp at 50°C.
I will try and see if there is another byproduct within it by TLC or if it is indeed toluene.
Using the azeotrope is a good idea. I'll try adding methanol and vacuum distilling it off before using it any further if that is the case.
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
Looks like there is indeed another product if I am not mistaken, separation is not very good with this solvent system ethylacetate:hexane and a drop
nh3
(unfortunately the best result of the ones I've tried).
chloroform:ethylacetate gives only one spot without tailing.
chloroform:ethylacetate i2 stained (left side) this gives only two distinct spots

ethylacetate:hexane:nh3 trailing a lot but I think there are two products visible?
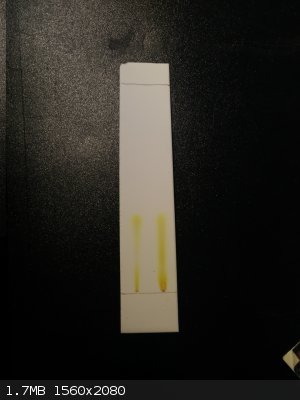
ethylacetate:hexane:nh3 i2 stained

[Edited on 27-2-2015 by lullu]
[Edited on 27-2-2015 by lullu]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
I'm guessing you're new to TLC. What ratios of solvents did you use?
Why add ammonia? Thats really not going to help here.
An additional note that may help me troubleshoot this with you... you started with p-methoxyphenol and were attempting to make
2-hydroxy-5-methoxybenzaldehyde, right?
[Edited on 27-2-2015 by DJF90]
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by DJF90  | I'm guessing you're new to TLC. What ratios of solvents did you use?
Why add ammonia? Thats really not going to help here. |
Yes I am.
it was 1:1 ethylacetate:hexane volume wise and one drop 10% ammonia, I added it because I assumed it might help to stop the trailing.
The other run was 1:1 too.
The TLC without NH3 ran very fast the two dots are in fact on the upper end of the TLC, it looks like it is quite pure on this one, the other TLC
looks like it is a mixture of two substances if I interpret it right.
[Edited on 27-2-2015 by lullu]
Yes the product I use for the TLC is in fact 2-hydroxy-5-methoxybenzaldehyde, I searched for vanillin TLC eluents but was not able to find some
recommendation with solvents I have at hand.
[Edited on 27-2-2015 by lullu]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Heres what I would do:
1) Your sample looks to be too concentrated, hence the large amount of streaking. I would take half of your sample solution and dilute it with an
equal volume of solvent (what is your sample made up in? DCM or chloroform is a good choice).
2) Take your TLC plate and mark three origins about 1cm off the baseline. Left one for starting material, right one for crude product, and the middle
for a co-spot (both samples spotted in same place). You probably need to make up a sample of your starting material for this - I'd try for something
like 5-10 mg per mL.
3) You mention that 1:1 EtOAc-Hexane runs almost to the top of the plate, and it looks like you have one species. This is because this solvent system
is too polar - aim to get your spots towards the middle of the plate: Rfs between 0.1 and 0.8 will do nicely. I'm going to guess that 1:4 EtOAc-Hexane
(by volume is convention) will be a good start.
4) Visualisation: If iodine is all you have on hand then fair enough. If your plates are F254 type, then fluorescence under UV (254 nm) would be a
good place to start. If they're not fluorescent plates, then you may still have luck using a long wave UV source (a blacklight bulb is good for this
and emits around 365 nm which is whats used in commercial UV lamps for TLC). If you have access to vanillin stain, I'd try that, too (instead of the
iodine...)
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
Sample might be indeed too concentrated, it is diethyl ether.
I just ordered a 254nm UV-S. I'll run it on monday like you described and let you know.
Thank you DJF90, I really appreciate your time and efforts.
[Edited on 27-2-2015 by lullu]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
I'll check back monday then. Feel free to send a U2U if you need help with anything else.
|
|
|
Chemosynthesis
International Hazard
Posts: 1071
Registered: 26-9-2013
Member Is Offline
Mood: No Mood
|
|
I'd like to add that you may be using too broad of spots. You might have trouble fitting that cospot in there between the two you have unless you draw
out a capillary or pipette tube over sooty flame and use that to spot your TLC. It helps with dilution as well, since a finer spot can afford to be a
little more concentrated.
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
The spotting was indeed too high concentrated.
Here is the result:
Left side is pure 4-methoxyphenol, middle is the product mixed with it. Right side is the product after isolation without distillation.
The product of my Mg ortho formylation (with good temp control around 98°C. the whole time) contains a small amount of 4-methoxyphenol and another
product besides 2-hydroxy-5-benzaldehyde it seems.
Very interesting 
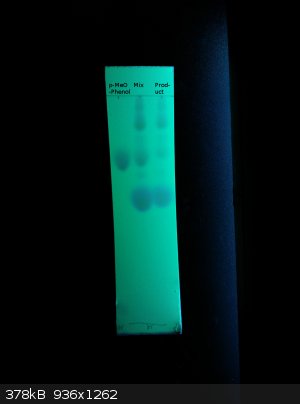
[Edited on 3-3-2015 by lullu]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
That worked much better, well done. What eluent are you using for that separation?
You've confirmed an impurity by TLC. Now, what are you going to do about it?
I have a couple of suggestions:
a) Attempt to recrystallise a small portion from whatever solvent system the literature recommends. Then you can TLC the re-crystallised product
against the crude and see if you've removed any of the impurities.
b) You could speculate that the spot with lowest Rf may be the benzoic acid (from aerial oxidation of the aldehyde). You could take a small amount of
your crude into ether and wash with sodium bicarbonate solution. Then TLC the ether layer (at an appropriate dilution) to determine whether the
impurity has been reduced/removed.
c) You mentioned distillation may be an option. Hopefully the other components are separable, but depending on scale you may have high losses due to
surface area etc. (What scale are you working on?)
d) You could column it. Its quite a solvent intensive method for purification but it does work, although you may want to practice a bit first.
e) Do nothing about it. If the impurities won't interfere in the next reaction (if there is a next reaction...) then you could use the crude and
perhaps achieve an easier separation at the next stage.
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
1:4 ethyl acetate/n-heptane
| Quote: Originally posted by DJF90 |
b) You could speculate that the spot with lowest Rf may be the benzoic acid (from aerial oxidation of the aldehyde). You could take a small amount of
your crude into ether and wash with sodium bicarbonate solution. Then TLC the ether layer (at an appropriate dilution) to determine whether the
impurity has been reduced/removed.
|
That is likely, although degassed and standing over argon it is a few weeks old now.
You think the diformylated product would be having a bigger Rf?
Would the bicarbonate wash not form a water soluble phenoxide with my intact phenolic aldehyde?
| Quote: Originally posted by DJF90 |
c) You mentioned distillation may be an option. Hopefully the other components are separable, but depending on scale you may have high losses due to
surface area etc. (What scale are you working on?)
|
Yes, distillation is not really an option at this scale, but if the second product is the diformyl product I will do it.
| Quote: Originally posted by DJF90 |
e) Do nothing about it. If the impurities won't interfere in the next reaction (if there is a next reaction...) then you could use the crude and
perhaps achieve an easier separation at the next stage.
|
Forming the bisulfite adduct after alkylation of the hydroxyl group and washing with NaOH solution might be the best method to get rid of the second
product if it is indeed the oxidated aldehyde.
I will try and oxidize a sample and rerun TLC against it.
Once again thank you DJF90.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Good to see I gave good advice.
| Quote: Originally posted by lullu | | Quote: Originally posted by DJF90 |
b) You could speculate that the spot with lowest Rf may be the benzoic acid (from aerial oxidation of the aldehyde). You could take a small amount of
your crude into ether and wash with sodium bicarbonate solution. Then TLC the ether layer (at an appropriate dilution) to determine whether the
impurity has been reduced/removed.
|
That is likely, although degassed and standing over argon it is a few weeks old now.
You think the diformylated product would be having a bigger Rf?
Would the bicarbonate wash not form a water soluble phenoxide with my intact phenolic aldehyde?
|
I'm not sure where the diformylated product would run. A bicarbonate wash should only remove any acid (if present) due to the appropriate pKa values
(its not a strong enough base to deprotonate the phenol to any great degree).
| Quote: Originally posted by lullu |
| Quote: Originally posted by DJF90 |
e) Do nothing about it. If the impurities won't interfere in the next reaction (if there is a next reaction...) then you could use the crude and
perhaps achieve an easier separation at the next stage.
|
Forming the bisulfite adduct after alkylation of the hydroxyl group and washing with NaOH solution might be the best method to get rid of the second
product if it is indeed the oxidated aldehyde.
|
If the impurity is the acid then a bicarbonate wash should remove it satisfactorily. Shouldn't be any need to form bisulfite adducts.
Try the wash with bicarbonate first. If the impurity isn't reduced or removed, then its probably not the acid.
If you have 2,4-DNPH use that to stain the plate. Aldehydes and ketones show up as orange spots on a yellow background - if that impurity stains too
then its suggestive of the diformylated product.
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
indeed!
| Quote: Originally posted by DJF90 |
I'm not sure where the diformylated product would run. A bicarbonate wash should only remove any acid (if present) due to the appropriate pKa values
(its not a strong enough base to deprotonate the phenol to any great degree).
|
Could you explain me how you estimate the pKa value of the 2-hydroxy-5-benzaldehyde?
| Quote: Originally posted by DJF90 |
If you have 2,4-DNPH use that to stain the plate. Aldehydes and ketones show up as orange spots on a yellow background - if that impurity stains too
then its suggestive of the diformylated product. |
sadly I do not have access to it at the moment, it certainly seems like a good investment.
I'll try to wash it with bicarbonate and report on the results.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by lullu | | Quote: Originally posted by DJF90 |
I'm not sure where the diformylated product would run. A bicarbonate wash should only remove any acid (if present) due to the appropriate pKa values
(its not a strong enough base to deprotonate the phenol to any great degree).
|
Could you explain me how you estimate the pKa value of the 2-hydroxy-5-benzaldehyde?
|
Well, phenol has a pKa of about 11. Methoxy group might increase that a little, but by far the dominant effect is that of the carbonyl group, allowing
delocalisation onto the carbonyl oxygen. This increases the acidity of the phenol. Fortunately I have access to a license key for MarvinSketch,
allowing use of the pKa feature:
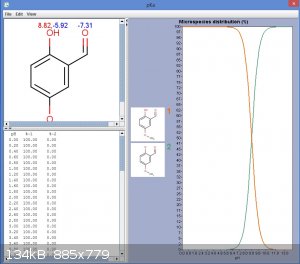
EDIT: Actually a bicarbonate wash might remove much of the diformylated product also. The predicted pKa came out at 7.8, which is similar to
bicarbonate.
[Edited on 3-3-2015 by DJF90]
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
Thank you so much!
|
|
|
| Pages:
1
2 |








