| Pages:
1
2
3
4 |
semiconductive
Hazard to Others

Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
I think I found another example.
Experiment #492, in order to get away from alkalai metals which can cause Kolbe reactions;
I want to try Ammonium Nitrate. This will not dissolve, though, in 7 drops of acetic acid and 21 drops of ester (or ether...,etc.)
It will dissolve in Formic acid, because of how small it is and similar to water.
I decided to mix up 7 drops of acetic, 21 drops of ester, and 1 drop of formic acid;
This ought to slowly dissolve a prill of ammonium nitrate.
Here's the experiment after running a few hours:

It has 5 [mA] flowing; which means the 7 drops of acetic acid I put in will be used up within 48 hours *if* the acid is attacking the nickel.
However, if you look closely at the cathodes; you'll notice the low current (left) cathode has a brownish coating on it; the higher current cathode,
though, is clean.
There's not even a whitish film on it.
I've seen this happen several times in the past; and often (not always) this indicates a reaction that will never stop. Sometimes this kind of
reaction will bubble gas for a few days, and then eventually stop gassing. But, the electricity keeps on flowing.
However, it never plates metal. But it often will show a build up of something on the low current cathode, exactly as seen in the picture.
I'm wondering if it's possible for metals to partially reduce at the cathode, say from Nickel ++, to Nickel +, and then return to the anode to be
oxidized again?
Since the + electrode is always repelling positive ions; Can anyone explain the mechanism that might make this happen?
I have cellulose acetate which is permeable to +ions, and can make a separated cell; but if nickel is changing from +2 to +1 oxidation state; wouldn't
it be able to pass both ways through the membrane? (Eg: a membrane wouldn't be able to force +nickel ions to stay in the cathode area and be fully
reduced to metal).
Any thoughts on techniques to break this kind of cycle?
Also, I notice, the prill of ammonium nitrate continues to bubble as if dissolving; but it's not getting any smaller. This definitely puzzles me.
[Edited on 1-12-2023 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Did some searching, and came across this:
Electroplating nickel, UK paper.
I'll have to see what I can learn from it. Although the Cyclic Volt-ammetry doesn't include sweep rate information for the plots; and the
electrode's surface areas are not known for sure.
[Edited on 4-12-2023 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Experiment #492 never plated. Fail. Continues to conduct electricity, with very slowly reducing currents.
Whatever gunk is on the low current electrode appears to be the only product.
Experiment #493.
Tackling the pH solubility question.
I know Iron, in water, is in the +3 form when pH < 4. From a couple of tests, I think it does the same thing as expriment #492. Everything turns
to rust/sludge in the water, and does not plate metal.
I know nickel, in water, does not plate at pH<3, from sulfamic acid in water. But it will at higher pH.
Even though nickel is only supposed to have a +2 oxidation state for bonds, the previous paper reveals that with Chloride ion present; it attracts 3
negative chloride ligands.
So, I'm under the impression that it's possible that nickel in a waterless solution, is dragging along more vinegar molecules than it would do in
water.
The normal way to solve this problem is to add a base to the solution, to buffer the pH.
Except that pure acetate doesn't ionize in the first place; but it does ionize in the presence of alkalai metals, or salts, which may be acting
(artificially) as a super acidic pH since the acid molecule is in excess. Adding a metalic salt, then, would be counter productive.
It occurs to me that another common buffering agent is an extremely weak acid coupled with an alcohol.
I know that having multiple exposed oxygens (in pairs) is enough to cause anode erosion, and ion conduction.
So, it seems to me a possible choice of ionizing substance is to use Boric Acid + a poly-alcohol, and reflux the mix until a solid ester is formed.
This might, hopefully, act to buffer the nickel solution to make it act *as if* it had a less acid pH.
7 drops volume of boric acid, refluxed with 7 drops poly-ol-alcohol, until very viscous/ nearly solid. Add to 1CC of pure acetic acid. Heat with
soldering iron, gently, until dissolved.
Nickel electrode +, 2 graphite electrodes -.
Initial test of conductivity is good; the Boric acid's oxygen + alcohol, does cause the glacial acetic acid to become conductive.
5 mA current flows. I can see nickel haze forming slowly in the first half hour of operation.
Solution is rapidly greening, which suggests the alcohol + Boric acid might be abnormally breaking the dimmer bonds and increasing the exposure of
free acetic acid to the nickel. ( It's not the boric acid, alcohol, by itself which is conducting the current; as the color is not green. )

This appears, even within three hours of starting, to be a successful plate; although it is bubbling hydrogen out at the cathodes.
There is un-evenness in the nickel plating; but that is normal when hydrogen gas is bubbling off the cathode. I'll have to wait to see if the
hydrogen ever stops bubbling or not.

[Edited on 5-12-2023 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
It plates nicely at first, but the conductivity of the solution continues to rise; 10x.
The current density gets high enough that nickel starts plating black-ish, and brittle.
As the liquid gets near to saturation with nickel, the hydrogen evolution slows down.
But, it stops plating at that point. Current drops due to the liquid viscosity becoming extremely high; and it's opaque.
I tried adding an ester to dilute the liquid, which made it semi-transparent and the conductivity did rise again; but it didn't resume plating. This
was a very interesting experiment. 
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
#494, mixed carboxylic glacial + ether + SO2 bearing molecule.
This is the pre-plate phase, where I'm just running a titanium anode at 5-10mA.
I boiled the SO2 bearing molecule in ether, in order to drive off any water of crystallization.
Here, I'm using titanium to get rid of any remaining moisture.
But, surprisingly, it's plating titanium/rutile mixtures !!
On the low current cathode, the plating is metallic. On the high current electrode, it's blackened; also the titanium anode blackens as well.

My choice of SO2 radical bearing molecule, I've been varying to see if it makes any difference. There's solubility differences; but chemically, the
same activity tends to happen when SO2 is present -- bubbling is suppressed on the cathode, and shifts to the anode.
I'm suspicious that the bubbling is "boiling" of the lightest carboxylic acid, because it refluxes on the glass of the test tube rapidly; and
condcutivity increases as drops of condensate fall back into solution. Even with solution cold, the test tube fogs within a minute of starting the
current. Electric ion boiling, perhaps ?
[Edited on 14-12-2023 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Experiment #496 Calcium Butyrate + Ammonium nitrate in 99.97% IPA.
This is a partial success.
My idea was simply that calcium might bond to the nitrate, and make the ammonium molecule freer to react in solution. ( eg: Nickel is less reactive
than calcium, ). Butyric acid salt was chosen because it's easy to get, and butyric acid being a similar acid to acetic; might also plate although
it's a lot weaker than acetic.
It didn't go as I expected, but it did go!
There was little or no dissolution of ammonium nitrate in this solution, at first.
But the conductivity rapidly rose and the color of the solution became a light green, with quite a bit of coloidal suspension.
It then began plating black material, dendriticly.
But each time I knocked off the black material, and heated the solution with a soldering iron, (and thankfully, it didn't explode -- outside. ), the
next layer of deposited material was lighter colored.
After one day, the chemical deposit is no longer dendritic; although still grey/black.
vi: 10mA on 10mm length of 0.9mm diameter graphite cathode.
This is a bit unusual, for I've tried sodium nitrite in quite a few experiments; and it usually doesn't plate anything. So, I wasn't expecting
nitrate with it's extra oxygen to work at all. But, it does.
The color, light green, I think means I've got nickel nitrate.
It must be *extremely* soluble in alcohols. Which means it doesn't stick to the electroplated surface.
I looked up nickel nitrate, and it is (indeed) used for electrotyping and forming in water baths.
I'll have to ignite some later, to find out how combustable it is. But this looks promising, if it's not too energetic.
Being as there is no water in the solution, I'll also be curious to see if I can crystalize out Nickel Nitrate by evaporating off an alcohol in the
presence of MgSO4 dessicant; with either IPA or methanol.
It might be a viable way to grow large nickel nitrate crystals, which is pretty impractical from water.
(NiNO3 . 6H2O, Is too deliquescent to form nice crystals when I tried it.)
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Experiment #504. See if Acetone can complex divalent metals, such as nickel, and/or plate:
1 CC non-protonating solvent; Eg: THF, DMSO, sulfolane, polaxamer, ether, etc.
Test for conductivity before starting experiment, no-conduction indicates good purity.
Add 7 drops of acetone to solvent.
Cap mixture with kerosene to reduce moisture absorbtion.
I applied voltage. Current begins flowing from Ni(+) anode to graphite (-) cathode. ~4 [mA] (in sulfolane -- nickel anode stays clean. )
~2.5[mA] (in DMSO -- note: blackens nickel anode. )
Surprising:
Initial bubbling forms in liquid near the cathode, but not on the cathode itself. This is less apparent the lower the initial current is, eg: DMSO
does it but not as strongly as sulfolane.
I can see the liquid mixing and going away from the cathode before bubbles form.
I'm not sure why/what this means. But perhaps the reduced hydrogen is initially soluble but negatively charged, and repelled by cathode? Curious!
Over night, solution turns a dingy yellow color. (Depends on the lighting, some angles look green others yellow.).
DMSO has the color but is more grey over-all; probably a side reaction.
This suggests that acetone can complex a divalent metal ion (transition metals, Ni,cu, etc. ) ( It's likely then that it will also work with
trivalent Fe. Future test! )
Current level decreases to 2 [mA], over-night, and bubbling stops. Current continues, but no sign of plating.
Add 14 drops of acetone. Bubbling re-starts, but this time it is physically on the lowest part of the cathode. eg: quite normal looking.

I can see a little discoloration of the nickel anode; so perhaps there is some solid reaction product possible, too. EDIT: No, it was just pitting.
The nickel is clean.
From studying acetone, I think that the alcohol/enol form of the chemical is less stable than the ketone form. I would suspect that this makes
hydrogen evolution harder, since a more stable alpha hydrogen needs to be displaced before an enol can be formed. I hope this will make acetone more
willing to give up complexed metal ions.
But: The hydrogen can be displaced/removed from the Enol form, and should allow acetone to chemically react with nickel: either one or two molecules
per nickel ion:
I suspect: [ C3H5O- ]2 [Ni++] (I am open to suggestions, or corrections; recall, I'm an amateur at best. )
I think: Positively charged nickel ions, then, can be complexed by acetone and/or form a salt with an acetone, enol tautomer, molecule.
I find it surprising that no sign of plating occurs with this solution even after hydrogen evolution stops. I'll clean the anode, and wait until the
second dose of acetone (+14 drops) stops evolving hydrogen.
This brings the acetone content up to around 50% of the solvent volume.
bubbling stopped several hours after re-starting and conductivity dropped to the micro-amp region. There's no way that all the acetone reacted, so it
seems to me that the yellow/green product might be suppressing further ionization of the acetone.
Pure acetone, 1CC, will conduct about 3micro-amps for a few minutes due to moisture; and then drops to zero. Acetone by itself isn't conductive. The
oxygen bearing solvents cause acetone to become conductive.
[Edited on 1-3-2024 by semiconductive]
|
|
|
Twospoons
International Hazard
Posts: 1349
Registered: 26-7-2004
Location: Middle Earth
Member Is Offline
Mood: A trace of hope...
|
|
Quote: Originally posted by semiconductive  | #494, mixed carboxylic glacial + ether + SO2 bearing molecule.
But, surprisingly, it's plating titanium/rutile mixtures !!
[Edited on 14-12-2023 by semiconductive] |
This is interesting - if you could get a plating of Ti4O7 that would be quite a valuable and unique accomplishment.
Helicopter: "helico" -> spiral, "pter" -> with wings
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
| Quote: Originally posted by Twospoons | | Quote: Originally posted by semiconductive | #494, mixed carboxylic glacial + ether + SO2 bearing molecule.
But, surprisingly, it's plating titanium/rutile mixtures !!
[Edited on 14-12-2023 by semiconductive] |
This is interesting - if you could get a plating of Ti4O7 that would be quite a valuable and unique accomplishment.
|
At my level, I'm just surprised to get any plating at all.
I don't know how to test which kind of titanium deposit I have on the cathode.
I've recently found a program molcalc, and have been drawing acetone in various configurations, to see if I can work out anything about which
molecules would be stable in solution and which ones wouldn't for experiment #504.
eg: I can get the energies of vibration (etc.) and compare that against a molecule with nickel replacing the hydrogen in the enol tautomer. I was
hoping I might be able to figure out a color for the ligand this way; but I am not there, yet.
The same ought to be possible with the titanium experiment, since that interests you.
I would be happy to re-run the titanium experiment in several different ways, and see what happens; but without some theoretical guidance from someone
experienced, I don't have much hope of solving the technical issues of getting a particular kind of deposit.
To get Ti4O7 requires a deficit of oxygen.
If that's a darker version of rutile, it may already be happening.
I don't know.
I have a fiber optic spectrometer (ocean optics), basic reagents, etc. but I lack the knowledge/experience to make definitive tests about what kind
of oxide is forming.
I would expect that whatever I have made (already) may be contaminated with sulfur as well. The anode blackens as well as one of the cathodes which
has deposits on it.
If you are willing to make suggestions, I would certainly try your experiments and post results. Care to gamble?
[Edited on 29-2-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
For example, I mocked up two acetone molecules in the ENOL form and joined them with a divalent atom. Nickel isn't available in MolCalc, so I used
magnesium.
But, this is one possibility for what is happening to the acetone in the presence of nickel.
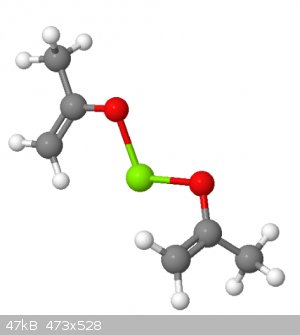
|
|
|
Twospoons
International Hazard
Posts: 1349
Registered: 26-7-2004
Location: Middle Earth
Member Is Offline
Mood: A trace of hope...
|
|
I would love to be able to make suggestions, but chemistry is just a hobby for me so my theoretical knowledge is sorely lacking. The Ti4O7 sub-oxide
is interesting because it is both conductive and fairly inert, making it an excellent anode or cathode coating material. Usually it is produced by
reducing TiO2 with hydrogen or carbon at high temp (900C+), and anode coating processes seem to involve things like thermal plasma spraying. If you
have stumbled on to a low-temp electrolytic process, then that is quite something.
Helicopter: "helico" -> spiral, "pter" -> with wings
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
I see.
Well, whatever it is on the electrode; it *is* conductive and it does contain titanium.
It fails the test for sodium, and it is semi-bright metallic shine on the low-current electrode -- although I still think it's an oxide. (It's not
pure titanium metal, whatever it is.)
You could replicate the experiment pretty easily using a piece of titanium wire.
I was attempting to dehydrate some reagant's water of crystallization.
Of the set I tried that day;
Sodium Saccharine is one that contains SO2 and is easy for anyone to get.
That particular experiment was a variation:
Ether eucalytis globulus (purified) or Tri-ethyl citrate as bought (both work the same), acetic acid, formic acid, as solvents.
1/2 cc TriEthyl citrate, 1/2 cc acetic, and enough Na-Saccharine to saturate it.
I boiled it with a 25 watt soldering iron in a glass test-tube until the saccharine was dry and steam stopped forming on the test tube (overnight);
then I tried adding formic acid (just enough) to make half the saccharine dissolve.
This resulted in a clear fluid, which after many hours, stopped producing vapors. Which means the formic acid that was left became stable.
Then I ran electricity from a titanium anode to two graphite cathodes;
It was a very crude setup since I was just trying to get titanium to go into solultion and chemically combine with any traces of water that were left.
I expected this to cause either precipitation, or to chemically bind the water to titanium which would then not affect nickel plating, since Ti is
more reactive than Ni. Either way, it would do what I wanted.
60HZ , 125 VAC power, through a diode, through a 4.7K resistor into a titanium anode and a pair of graphite electrodes; one hooked directly to
neutral, the other hooked through a resistor picked to reduce the current by about a factor of 10. ( I didn't write the value down. )
You're welcome to replicate it.
It's not my interest in this thread; but I'm happy to share any info, publicly as I stumble across things. 
[Edited on 1-3-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Experiment #506.
I can't get NaCl to dissolve in acetic acid; but I notice that MgSO4 (epsom salt) is used by a few people successfully in water with vinegar.
So, I want to see if I can plate with mostly crystalline epsom salt + glacial acetic acid, diluted with an ester or ether.
I put about 1CC of "Dr. Teal's" epsom salt from the store into a test-tube. I expect this is contaminated with small amounts of copper, mangneese,
and other metals. But, I can get reagant grade, later, if this works.
I added just enough glacial acetic acid to cover the MgSO4 crystals. Then I cap the experiment with 2CC's of inert kerosene.
My hope, here, is that the MgSO4 will tightly bind it's water of crystallization, but still dissolve in the acetic acid.
If it does, then during plating a certain amount of water of crystallization will be destroyed as hydrogen gas; and hopefully, whatever tiny amount
remains will not want to separate from Mg into solution; eg: this might make a 'nearly waterless' electrolyte.
Nickel+ graphite-.
I added 1 drop SO2 bearing molecule to cause initial ionization.
Initial bubbling is intense as expected. After about a half hour at 10mA current on 1cm of 0.9mm diameter graphite; I can see silvery-coppery looking
metal plating onto electrode. So, it does work (at least at first.)
After running several hours, I can see the epsom salt is dissolving into the glacial acetic acid. But it's becoming jelly like.
So, I add a few drops of ester or ether to dilute it.
Over-night, bubbling completely stops. Conductivity dropped to almost nothing, but it's easy to see the entire tube has become a gel again. Quite a
bit of metal did plate onto the graphite. I replace graphite with a fresh one. Add 6 drops of acetic and stir.
Solution becomes liquidy but colloidal.
The new graphite appears to be plating (slowly). Most of the colloidal suspension falls to the bottom of the test tube as the solution becomes
warm/hot.

There is much less bubbling this time around. I think this experiment is going to be successful. I need to figure out how much epsom salt is needed
for a given amount of glacial acetic acid in order to make a final solution with minimum colloidal solids remaining. I think it's an optimization
problem. Cool!
After 48Hours, pretty much the only place bubbles come from is the contact point between the graphite and the epsom salt. Bubbles are hardly forming
(at all) in the liquid areas on the visible part of the graphite electrode. The current level has dropped, considerably, though. From 10mA
yesterday, down to 2mA today. I'm going to allow it to run at 2mA for a full day and see if the plating properties change.
This picture a fresh electrode, that has been electroplating for 20 minutes at 2mA.

You can see: The liquid is still electroplating even in places where bubbles are not being formed, and the color of the deposit is about the same as
at the start of the experiment.
It's not a normal nickel/silverly color, but is tinted salmon pink to brownish as if some copper were alloyed with it.
This is a bit puzzling. Nickel sulfate is bluish, Nickel acetate is greenish, and neither of these colors are showing as a tarnish on the plated
metal;
There's likely a tiny bit of contamination metal in 1CC's worth of epsom salt. eg: But only something like 0.01%. That should have already
deposited out by now!
I would expect to be getting a nickel plate at this point.
It makes me wonder if I've got some kind of magnesium-nickel alloy plating, or maybe a basic hydroxide. I'm not sure what color those would give.
Google(TM) searches don't help much.
[Edited on 5-3-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Experiment completed: The color does slowly change to silvery nickel as the reaction proceeds. The current continnues to drop as the reaction
proceeds; after 96 hours, it was stable at 1.8 mA (regardless of voltage applied.)
I think this means that the ionization process with acetate/MgSO4 is pretty close to independent of temperature and voltage.
The reaction is too slow to do electro-forming, or thick nickel plating. It could only be used to make a thin surface coating of nickel. But, it does
work.
Noticeably, the MgSO4 prevents glacial acetic acid from becoming emerald green and absorbing a large amount of Nickel. But, it also increases the
percentage of Nickel that plates from solution per coulomb. This suggests that like pH problems in water, that the concentration of Mg ions will
affect the plating quality and rate, significantly.
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Experiment #507
See if conductivity can be increased by using combination of formic acid (stronger), and propionic acid (weaker), than acetic.
Start: 1/2 cc roughly of MgSO4.
Cap with Kerosene.
Boil with 25watt soldering iron for about half an hour. Attempting to dehydrate MgSO4 partially by getting rid of only excess water of
crystallization / water that is extremely weakly bound. I hope this will cause experiment to take less time to run without changing final results.
Fill with Formic acid until 1/2 cc space above MgSO4 is formic acid.
Nickel+, graphite-
Run for 48 Hours.
Current is notably less than many experiments I've done in the past using formic acid alone.
Green crystals form on the graphite electrode, and have to be removed many times.
As water content drops, the amount of crystals forming on cathode decreases. Eventually it becomes a non-problem, after several cleanings.
At this point the MgSO4 is colored a bluish-green, uniformly, and tends to become thrixotropic/solid when allowed to settle.
I decanted off remaining formic acid by slowly turning test tube and allowing liquid to drain. All green material stuck well to the bottom of the
test tube. This ought to be nickel-diformate and formic acid crystallized MgSO4.
Add propionic acid (which normally will not conduct electricity by itself).
Conductivity is very good (30mA), which is likely due to Formic acid dissolving in propioninc acid.
Lots of hydrogen bubbling (undesirable).
Picture after 12 hours of current applied to propioninc mixture.

Experiment hasn't been run long enough.
I think that's a crystal film on the graphite electrode, it doesn't appear to be quite metallic nickel. Maybe nickel propionate/formate mixture.
Hydrogen bubbling continues, current is still quite decent >10mA.
Will let run over the weekend, and will clean graphite electrode a few times.
Interesting how the nickel formate in the salt has become hetrogenous, with dark lines of Ni-carboxylate showing up an arbitrary distance away from
the electrode. I'm not sure why that's happening.
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|

Obviously a crystalline precipitate is sticking to the electrode (-), this March 11th,2024 morning. No signs of Nickel plating Metal.
The line of material that was greenish in color, 48 hours ago, is still green but it looks like it's darkened (concentrated) a lot. Compare the
pictures. I'm puzzled.
Conductivity has dropped, It was only 100 uA, this morning. As I cleaned the electrode, scraping off a hard crystaline deposit / perhaps an
oxide/hydroxide, I notice that the epsom salt continues to bubble even without the electrode present.
After cleaning, reinsertion of the electrode shows 0 uA current (immeasurably small.). Bubbles resume forming on the graphite electrode, though, so
there is a current at least in the liquid if not in the attaching wire.
I'm really not sure what to think about this experiment.
It's only a small deviation from the previous experiment, but it seems a total failure.
Oh, one detail. Propionic acid wouldn't evaporate in these conditions, but the liquid film is not visible. The current loss may be due to absence of
liquid solvent. I'll add a few drops (7) more propionic, and see if any change.
edit: +7 drops, still couldn't see film; it seems to mix with the kerosene a little too easily/abnormally. Added +8 drops more, then gave up. I can
see diffraction swirling in the kerosene, so maybe propionic acid will settle out in a few hours.
I checked the nickel anode; it had completely dissolved below the epsom salt layer.
I replaced it with a new piece of nickel; current resumes at 400 uA.
Edit 2:
Conductivity dropped again over a period of hours, no settling of the propionic acid is visible. I took a glass stir rod, and tried to stir the
epsom salt dust, but it had aggregated. It was hardest near the graphite electrode, and softest near the nickel source. I pressed down hard, and
was able to break it up again. Current rose to 4mA for a while, and then dropped back down to about 2mA.
I don't know if it's thermally induced, or a chemical reaction near the graphite; but on the glass, on the graphite, anywhere near the cathode;
crystals seem to grow rapidly in response to electric current flowing.
After breaking up the salt with a glass stir rod, I could see a meniscus about 2mm above the salt form. So the propionic acid is separating out now
that I've homogenized the epsom salt (again.). It's hard to see in photo, but above the meniscus uniformly across the test tube is a haze of crystals
in the kerosene sticking to the glass. Just below that; Where the propionic acid accumulates, the crystalline dust mostly washes off glass.


Note: I did not shake or stir the test tube between these two photos. They are with an hour or two of each other. The bubbles inside the epsom salt
of the upper photo have expanded and knocked the salt around. Gas forms inside the salt, near the electrode. I suppose the earlier photos of the
dark band, might be a channel where tiny gas bubbles formed and it's shape is just a matter of random luck.
The color is lightening, slowly, in the salt. That would suggest either dissolving of the darker green nickel formate from before, or a chemical
reaction of some kind. The current level is staying low, so I think it's more likely a reaction than dissolution of a conductive salt. Hmmm... it's
not headed in the correct direction to ever plate.
[Edited on 12-3-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Rough calcluation:
After doing several tests, the glacial acids tend to transfer as 48 drops for 1 CC of liquid with my pipettes.
Glacial propioninc acid = 0.9977 [g/cc] = .9977/48 = 20.78 [mg] per drop / 74.08 [g/mol ] = 280 [uMol] per drop.
Glacial formic acid = 1.22 [g/cc] = 1.22/48 = 25.4 [mg] per drop. / 46.025 [ g /mol ] = 552 [ uMol ] per drop.
Each drop of acid, then, ought to require a certain quantity of electrons to force all acid molecules to undergo a reaction:
Formic: 552 [uMol ] * 96485.3399 [C/mol ] = 53.26 [C] per drop
Propionic: 280 [uMol] * 96485.3399 [C/mol] = 27.02 [C] per drop
At 10 [mA],
1 Drop of propiconic takes 2702 [s] to fully react. ( ~0.75 hour/drop).
1 Drop of formic 5326 [s] to fully react. ( ~1.5 hour/drop.)
I use between 7 drops and 1CC of liquid in my tests;
Propionic runs between 5.25 to 36 Hours. (1.5 day maximum experiment run time. )
Formic runs between 10.5 to 72 Hours. (3 day maximum experiment run time. )
This means the present experiment ought to have already run to exhaustion.
It still has (after homogenizing) at about 2mA of residual current.
I add 1/2cc of formic, which brings the current level up to 10mA. Hopefully, the salt will turn darker green within 3 days. (Post will be below!)
There is enough formic acid to fill about half the salt's volume with a darker green nickel salt.
[Edited on 12-3-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
I've waited long enough to exhaust the 7 drops of formic acid that I added. After a week of electrolysis, there is no more of the green lines forming
in the powder. The powder has an aqua-marine, or jade hue. Occasionally, a very dark green powder will form on the anode; but after shaking or
stirring the powder, it re-dissolves.

You'll notice that the propionic acid has a greenish hue (looks golden yellow when viewed with eyes.) So, something is now dissolving into the
propionic acid which was clear in all the earlier photographs. But: The reaction/solution does not appear to be stable, as I can see color changes in
the solution which will precede different kinds of plating activity.
Yesterday night 12 hours ago, there was a silvery black deposit forming on the electrode. It was partially metallic. But, then the solution's color
changed and it rapidly began forming crystals of the epsom salt/nickel formate. It's as if the solution turned slightly colloidal, and
electroplating causes particles floating in the solution to adhere/precipitate on the cathode and (also) to the glass of the test-tube near the
cathode. It's electrically induced crystallization.
This is a very interesting experiment. The conductivity does not drop to zero when the acid charge is basically depleted. Each time I add an acid
charge, there is more conductivity after the formic acid is used up. The first time conductivity dropped to 500uA; The second time conductivity
dropped to 2mA; and after this last charge of 7 formic acid drops -- the current drops to 3 to 4mA. (3 with crystals on electrode, 4 when electrode
is clean.)
I'll see how much nickel I can get to dissolve into the solution and epsom salt, will post results below. I think nickel salts (double salts,
perhaps?) are increasing the conductivity. Propioninc acid isn't conductive by itself, but it does appear to dissolve nickel formate / nickel sulfate
/ whatever I have in there.
[Edited on 17-3-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
#507 is a very frustrating (and long running) experiment. The color of the salt slowly darkens (greenish) with time.
Often the solution will show color change from yellowish to greenish, and then suddenly will plate a black coating.
After that the current drops and plating stops for a while (just when your hopes are up that plating has begun!).
I've figured out that currents higher than 8mA are vaporizing the acids. To get maximum nickel dissolution, it's necessary to keep the current below
8ma on 1 inch or more of nickel.
I'm letting the experiment sit, and seeing if dissolution happens slowly from the salt. Perhaps I can get it to plate black a few times in a row, by
just letting it sit for a few days between attempts.
On the bright side ... I've found amidobutyric acid can be dissolved in small amounts of propionic acid & many different esters / ethers.
Today's picture is aluminum going into solution and trying to get rid of water before attempting nickel. As the reaction proceeds, the color of the
liquid becomes less colloidal and more clear/golden.

I see a very thin but noticable aluminum / perhaps oxide film on the graphite; although no metallic buildup. At higher current rates, the film is
more obvious in the kerosene area ... but etches back off with heating of the solution.
[Edited on 29-3-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Well lookie there!
It's aluminum plating near the tip (only) of the electrode in a 4-aminobutyric acid (not amido-sorry about the typo in previous post) solution. I
haven't even added a nickel anode or bromine, yet. This is still the moisture destroying phase of the experiment before I actually intended to run a
metal plating test. I've never seen this happen before!
Usually I centerfuge out the aluminum hydroxide, and then put a nickel anode in the tube.
The plating been getting better, but only at a certain depth in the solution.
Notice the very bottom of the electrode is not plating, and nor is the region close to the kerosene.
(You can see the hydroxide, so I think maybe a significant amount of moisture or oxygen is still penetrating the kerosene layer which is meant to
reduce moisture, sigh.)
I bought a compressed gas tank to supply an inert shielding gas. I'm going to try drying CO2 gas with a dessicant made specifically for that task,
and then slow flowing the gas to prevent atmospheric oxygen getting into the test tube as air or water. Hopefully, I'll have that working in a week
or so.

[Edited on 2-4-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
I'm still waiting April 2024, for CO2 tank parts. When I get the correct safety rupture disks, and a stainless mesh to hang sorbead blue dessicant
inside the tank with; I'll take photos. Anyone work with sorbead before? The stuff looks like silica gel with blue dye in it. I just got Jupiter
Pneumatics (TM) stuff off ebay.
Hopefully, I can bake it in an oven hang it inside the CO2 tank, and it will wick up traces of moisture in the CO2 that's put into the tank.
Today's shot is again of the preparation phase. I threw out the last test tube, and setup for a retry. This time I'm using butyl buterate as the
solvent (which, unfortunately miscs significantly with the kerosene), 4-Gaba & and aliaphatic hydrocarbon plus a prill of solid iodine for ion
carriers.
Iodine dissolves in the aliphatic hydrocarbon, which dissolves in the butyl buterate, quite well. Normally, literature i've seen online suggests
using Bromine to plate aluminum. But considering I got it to plate a little bit with no halogen at all, I decided to try a safer/cheaper iodine and
see if it improves the plating. Normally, by the 24 hour mark, iodine color would have lightened and visibility in the tube would improve. This
isn't normal.
But the answer, is *yes*. Iodine and/or aliaphatic hydrocarbon, and/or butyl buterate imrpoves the aluminum plating.

The original aluminum anode is most visible all the way to the right, against the glass, and is darkened with iodine. The bright silvery thing, just
to the left of it, is an electroplated graphite electrode.
If you notice the tip, it's not plated the same as the whole body. This is the same issue that happened in the last run. It looks like a hexagonal
set of dendrites on the tip with some graphite exposed.
The adherence of the aluminum in the last experiment to the graphite was not very good. I could slide an aluminum oxide ring off the graphite. In
this one, there is a thick enough plate that it should be more difficult to get off. But, it still might be contaminated with organics heavily (don't
know.) There is *much* less hydroxides/precipitates in this experiment than the last one. Current is on the order of 3 mA at the start, and 800
micro-amps this morning. Still, for a second try, I couldn't ask for more.
Until I get Argon or CO2 shielding gas, I don't really have a way to completely stop oxide formation.
[Edited on 8-4-2024 by semiconductive]
|
|
|
Rainwater
National Hazard
Posts: 987
Registered: 22-12-2021
Member Is Offline
Mood: Break'n glass & kick'n a's
|
|
This one is on a mission. Im rooting for you dude/dudette.
| Quote: | | traces of moisture in the CO2 that's put into the tank. |
Not sure how big your tank is but if you can bring it to <0c, the reduction in pressure from usage will keep any water frozen solid and should
provide a nice dry stream. With 2 containers, and a compressor. This can be done in batches, for use later.
[Edited on 9-4-2024 by Rainwater]
"You can't do that" - challenge accepted
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Experiment #510
A nice separation,
I've been trying to make alkoxides (strong) to attempt plating in.
In general, I've been attempting to use K-OH and electrolysis, to make a progressively stronger and dryer alkoxide.
But; most solutions turn super dark brown or black during the process.
I tried 1,3 propane-diol, glycerol, and di-propyl-diol (food grade, O bond in center.). All of them turn brown (or reddish-brown) during
electrolysis.
I've also noticed that formic acid tends to have a brown color after exposure to air/light for a while. I'm not sure if this is oxidized chemical, or
if it's an impurity. But, since it makes the experiments opaque; I decided to look for ways to remove the color (if possible).
In this experiment, I used ~1 cc K-OH crystals, then I added enough 1,3 propanediol to fill all pores and cover the crystals.
Then I added 1 CC kerosene, as an inert cover layer in the test-tube.
Since dissolving KOH is a problem, and tends to leave chunks and solidify during electrolysis; I decided to use a few drops of formic acid, to bring
the bulk of the potassium into a liquid solution. I figure that the water formed, and possibly the K+formate, can be removed by electrolysis.
I heated and stirred, and slowly added just enough formic acid (drop by drop) to cause almost all the KOH to dissolve, but leaving a trace of it.
Then I added a few drops of poloxamer-124. This is much less dense than the potassium/diol/formate solution and slightly soluble in kerosene. I was
hoping it would mostly float between the kerosene and alkoxide mix; it does (very well!)
Then I placed a graphite cathode, with an insulating sleeve except for the very tip, into contact with the diol/KOH solution; and I placed an anode
*above* the poloxamer 124 layer by about 0.5 mm. This is close enough that ions in the poloxamer will penetrate the kerosene and hit the anode. The
anions then electrolyse to release oxygen (from water). I think formate ought to turn into some kind of gas (An alkene or CO2, Kolbe electrolysis;
this is not an acidic electrolyte, but it's CO2 when acid as shown by Woelen: https://woelen.homescience.net/science/chem/exps/precision_e... ).
This result appears to be 4x (or more) gas bubbles at the cathode compared to the gas at the anode.
As the reaction proceeds, the slight brownish color in the KOH/diol layer migrates *up* into the poloxamer 124 layer.
This does clarify the KOH/diol layer. Heating the glass test-tube with a soldering iron speeds up the color migration and clarification a lot. It
doesn't cause re-browning, either.
I've run it for 24 hours, and the KOH/diol layer is very nearly clear; with the poloxamer layer increased in width (about double original) and turning
medium brown. It's a very successful separation. This should (I'm hoping), allow a build up of alkoxide at the bottom of the test tube.

I'll allow this to continue to run; and see how it proceeds.
The current is resistor limited to roughly 30mA or less.
[Edited on 24-5-2024 by semiconductive]
|
|
|
semiconductive
Hazard to Others
Posts: 326
Registered: 12-2-2017
Location: Scappoose Oregon, USA.
Member Is Offline
Mood: Explorative
|
|
Experiment #510; postlog.
The use of poloxamer to separate our darkening electrolysis impurities during the addition of KOH, works reasonably well.
Electroysis of the resulting mixture for about a week with a graphite anonde, caused the solution to become a very light amber color which could not
be removed by more poloxamer. Still, the solution is clear enough to see the effects going on, which was my goal. The graphite electrode was
oxidized at the tip, and I suspect this is the source of the amber darkening that poloxamer could not remove.
I switched to a nickel anode and attempted plating for a week. The solution turned green, slowly, which suggests that Ni + a di propyl alcohol, or
potassium makes this color.
No metal plating was observed (at all). But, there was a continuous whitish crystalline formation on the electrode. Conductivity slowly dropped
over a period of a week.
I wonder if this could be K2O, since I'm destroying water; and I would have expected nickel to remain green. Anyway, it's run nearly to exhaustion,
and the green is strong enough that it's getting difficult to see the electrode; I could add ester and centerfuge in order to make it transparent
enough to watch again; but the conductivity would be very low and there's no sign of metal deposit which I would have expected by now; so I'll give
up.

|
|
|
bnull
National Hazard
Posts: 596
Registered: 15-1-2024
Location: Home
Member Is Offline
Mood: Sneezing like there's no tomorrow. Stupid cat allergy.
|
|
Your persistence (and curiosity, of course) amazes me.
| Quote: Originally posted by semiconductive | | But; most solutions turn super dark brown or black during the process. I tried 1,3 propane-diol, glycerol, and di-propyl-diol (food grade, O bond in
center.). All of them turn brown (or reddish-brown) during electrolysis. |
About 10 years ago, I tried to plate copper from a complex copper-glycerol-sodium hydroxide using lead and copper electrodes. Instead of a nice coat
of copper, what I got was a brick-red sludge of cuprous oxide that smelled funny. I'm not sure what it was (I didn't research) but reminded me of
cookies.
One solvent that you can try, if you haven't tried it yet, is molten urea. A large number of salts is soluble in urea slightly above its melting point
(~130 °C, see attached paper).
Attachment: R. E. D. Clark - Urea as Solvent.pdf (353kB)
This file has been downloaded 185 times
Edit: I forgot to mention that I had tested copper oxide in molten urea two months ago. Copper oxide dissolves with a slight fizz and
the solution turns blue. The conductivity is good enough to allow plating.
[Edited on 11-6-2024 by bnull]
|
|
|
| Pages:
1
2
3
4 |
|








