| Pages:
1
..
12
13
14
15
16
..
18 |
NeonPulse
Hazard to Others

Posts: 417
Registered: 29-6-2013
Location: The other end of the internet.
Member Is Offline
Mood: Isolated from Reality! For Real this time....
|
|
Yes. I can say that I do. I made Hg azide in tiny amounts in plastic vessels. I used Mercury 1 nitrate as this is supposedly much safer than the 2
salt. It detonates violently with an electric blue/white flash and is very sensitive to both impact and sparks. If made with HgNO3 2 you risk
extremely sensitive crystals that can detonate even from falling to the bottom of the solution it's contained in. I have not had a small detonating
crystal in the solution I had made.. I have a video HgN3 on the tube.
|
|
|
a nitrogen rich explosive
Banned troll
Posts: 176
Registered: 28-3-2016
Member Is Offline
Mood: Repentant
|
|
The mercury I nitrate version is extremely stable compared to the II version...
I think that Derek Lowe wrote about it on his blog 'Things I won't work with.'
I can't think of a better signature.
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Quote: Originally posted by NeonPulse  | There's also a lithium/copper azide complex I have read about that sounds interesting too. Lithium azide presents a challenge though without
hydrazoic acid, and I'm not sure the synthesis of this is something I'm willing to do. It's possible though to make it in a solution of ether or
chloroform but again the volatility... A 10% or less concentration is relatively risk less though
|
Simply change NaOH or KOH for LiOH in the normal NaN3 or KN3 process from Alkyl nitrite hydrolysis upon contact with hydrazine...
This is interesting lithium n-azidocuprates (n= tris, tetra, penta, hexa,...?)...so some transition metals azide may form (probably because of the
pseudohalegen behaviour) such complexes as hexacyanoferrate/-ite...
--> hexaazidoferrate of K?
K3Fe(N3)6
--> hexaazidoferrite of K?
K4Fe(N3)6
[Edited on 29-4-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by NeonPulse | It does have a pretty bad rep but I I found the copper II azide to be less sensitive as I first thought.it really is made out to be a monster. That's
not saying it is safe but It was still quite spark and impact sensitive but I'm not so sure about friction. I could not get it to fire when I ground
a small amount between steel surfaces, but I'm sure if I had of added glass or something else gritty and then rubbed it it would have likely fired.
The copper I azide seemed far more sensitive,easily detonating violently even with light taps of the hammer. Some of copper azide complexes do sound
interesting and I would like to try some of these if I can find a decent synthesis for them. I know the tetraamine copper azide can be made by running
a stream of dry ammonia gas over the Cu 2 azide or by dissolving it in ammonia and then evaporation of the liquid with the blue precipitate being the
product. I do know that it does quickly lose the ammonia though and reverts back to copper azide.
|
I am certain I have seen patents for rimfire primer compositions that deliberately used copper azide so it would seem that under precise conditions
for synthesis it is possible to make a form of copper azide that has predictable properties.
I have made the material but it has been 40 years ago and I observed the same low solubility that produces a colloidal precipitate similarly as for
lead azide when strong solutions of precursors are reacted.
A better but larger crystalline form can be obtained as a general method where dilute solutions are mixed by special technique, which involves
simultaneous addition of the two solutions of precursors, equal diluted volumes containing the amount of theory needed for each precursor being added
at equal rate. Metering pumps are ideal but two addition funnels adjusted to a slow equal drip rate can substitute.
I.V. solution bottle setups are ideal for this kind of simultaneous addition, and use the needles immersed into the liquid instead of letting drops
fall into the liquid. The drip rates can be visualized in an observation chamber on the manual old fashioned I.V. type, or an infusion pump can be
used and the delivery rate is digital display.
Anyway the gradual nature of the addition in a stirred liquid flowing past the tip of a cannula seeping in reactants swept into the current of the
stirred reaction will favor larger crystal formation. The outlet is immersed in the current of the swirling mixture, and a capillary with a small
opening or a gas dispersion tube like the aquarium bubbler diffusers can also be used as outlets, but the flows have to be established before
immersion and diffusers can become clogged if there is not a sufficient flow rate to keep the precipitate completely external and not migrating
upstream into and clogging the diffuser.
One of the streams of precursor should be introduced through a tube at a greater depth than the other on the opposite side of the beaker which
contains an amount of plain water that is about equal to the sum of the very dilute concentration precursor volumes to be added to the stirred
reaction, with slow addition streams and the reaction mixture heated and stirred. Such a scheme should produce larger but still microcrystals that
should filter quickly.
What is the limiting size of crystal development that may be a danger zone is unknown.
Anyway if you are experimenting with survivably small quantities and are curious this would be what to look at as interest is what changes with
increasing crystal size about the properties.
The more dilute the precursor solutions and the higher the temperature and longer the time of additions into the continuous stirred mixture, the
larger the crystals should grow.
The dilutions are pretty extreme that are used for these low solubility product systems where a gradual clouding and gradual crystal development is
forced to occur by conditions and method, and by extreme I mean for example a couple of grams of end product forming over many hours in a reaction
mixture totaling 4 or 5 liters at completion. If spontaneous detonation at a certain size of crystal development occurs, then of course conditions
have to be adjusted to use more concentrated precursor solutions, or lower temperature and faster addition rates, or all in combination.
It is possible to design "unvarying continuous reaction conditions" where the composition of the reaction mixture does not change over the course of
reaction and such technique should produce a uniform mesh size of crystal product, but finding what is optimum combination of conditions required is
the goal there and will lead to a possibly patentable process for the technician who works up those figures and claims the process as proprietary. If
there is a dollar to be made from the work, send me a nickel so I can go buy a clue, as my clue supply seems to be dwindling with age and I have fewer
and fewer of them to spare as time works its magic on me 
I have been thinking that Cu(N3)2 might be a good candidate component in mixture with p-DDNP or NHN as a kicker for those slower self accellerating
materials, maybe to produce a good cheap green energetic initiator mixture.
[Edited on 4/30/2016 by Rosco Bodine]
|
|
|
NeonPulse
Hazard to Others
Posts: 417
Registered: 29-6-2013
Location: The other end of the internet.
Member Is Offline
Mood: Isolated from Reality! For Real this time....
|
|
Rosco, did you have a patent number for that or is it one of them things that " those skilled in the art" type things? Your idea actually sounds like
a very sound method and would work very well I think. I had a quick look through some patents and only one I found mentioned using CuN3 in its
mixtures but it also said that the crystals were kept small using gum Arabic or dextrin. They never said how the azide was formed though. Maybe making
larger crystals of CuN3 is not such a good idea....
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
US5610367 cupric azide rimfire primer
is one patent and there is an application for a center fire primer formulation but I don't see any patent ever actually issued on that
Attachment: US5610367 cupric azide rimfire primer.pdf (760kB)
This file has been downloaded 907 times
|
|
|
Etanol
Hazard to Others
Posts: 188
Registered: 27-2-2012
Member Is Offline
Mood: No Mood
|
|
Hi guys. Here is another unusual method of preparation of AgN3 from medical Isonicotinohydrazide (C6H7N3O)
1 mol AgNO3
1,2 mol of C6H7N3O
about 2,2 mol HNO3
1,2 mol NaNO2
distilled water
1.
R-CO-N2H3 (3% solution) + AgNO3+ 2HNO3 = R-CO-N2H3*2HNO3+ AgNO3
(With shortage of HNO3 the reaction goes:
R-CO-N2H3 + 2AgNO3 + 2H2O => R-CO-N2H3*2HNO3 + 2AgOH )
2.
R-CO-N2H3*2HNO3 + AgNO3 + NaNO2 (10% solution, 20-25C, addition for 1 hour+exposure for 20-30 hours in a covered glass) = R-CO-OH*HNO3 + AgN3 +NaNO3 +
HNO3 + H2O
Yield is 85-95% of the AgNO3
AgNO3 (unreacted) + NaCl = AgCl + NaNO3
Have a good synthesis!)
[Edited on 28-5-2016 by Etanol]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Interesting .....do you have a reference for the synthesis ?
This would tend to support earlier speculations that other hydrazides could serve as useful precursors for azides.
Isonicotinic acid hydrazide CAS 54-85-3
synonyms Isoniazid also Pyridine-4-carboxylic hydrazide
Cas No.: 54-85-3
Molecular Formula: C6H7N3O
Molecular Weight: 137.14
Appearance: White to off-white solid
Molecular Structure:
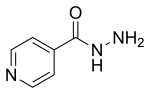
|
|
|
Etanol
Hazard to Others
Posts: 188
Registered: 27-2-2012
Member Is Offline
Mood: No Mood
|
|
Source is the lab journal of my friends 
Isoniazid is the most available organic acid hydrazide. The tablets are very difficult to filter. The easiest way is to use solution for injection.
Other hydrazides will probably require different pH and go mit a different rate.
Pb(N3)2 is impossible under these conditions due to reactions:
Pb(N3)2 + 2HNO2 +2HNO3 => Pb(NO3)2 + 2N2O +2N2 +2H2O
HN3 + HNO2 => N2O +N2 +H2O
May be this method also suitable for Cu(N3)2
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
A note of caution ─ copper azide is dangerous to handle and its prep. is best avoided!
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by Etanol | Source is the lab journal of my friends
Isoniazid is the most available organic acid hydrazide. The tablets are very difficult to filter. The easiest way is to use solution for injection.
Other hydrazides will probably require different pH and go mit a different rate.
Pb(N3)2 is impossible under these conditions due to reactions:
Pb(N3)2 + 2HNO2 +2HNO3 => Pb(NO3)2 + 2N2O +2N2 +2H2O
HN3 + HNO2 => N2O +N2 +H2O
May be this method also suitable for Cu(N3)2 |
See if you can get the name of that lab journal of your friend, and the issue and page numbers.
The discussion about use of hydrazides as a precursor for azides is something I had speculated in another thread that was off topic there should be
imported here. The post where I mentioned first the salicylic acid hydrazide is here linked, but that post needs to stay also with the other thread.
http://www.sciencemadness.org/talk/viewthread.php?tid=389&am...
Note to Bert or other moderator:
Everything from this below linked post and to the end of that thread should be imported to here in this thread
http://www.sciencemadness.org/talk/viewthread.php?tid=389&am...
The reference to salicylic acid hydrazide from the post preceding needs to be available here also in this thread and I will put a note in the pruned
thread linking to discussion of the hydrazide and azide here.
[Edited on 5/29/2016 by Rosco Bodine]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
It is not speculation, it is a fact known back to the early 1902...with Curtius
See Google Book: Peptides: Synthesis, Structures, and Applications - Bernd Gutte - p3-4

I have read it elsewhere... as a first method of making NaN3 (as explained by me a little below Rosco's post into Rosco's link)
This is very consistent with the reactivity of Ar-CO-NH-CH2-CO-N3 (hippuric acid azide) being comparable to acid chlorides...
Ar-CO-Cl + H2O --> Ar-CO-OH + HCl
Alk-CO-Cl + H2O --> Alk-CO-OH + HCl
so
Ar-CO-N3 + H2O --> Ar-CO-OH + HN3
Alk-CO-N3 + H2O --> Alk-CO-OH + HN3
And with basic catalyst/media one ends up with carboxylate and azide of the base cation
Ar-CO-N3 + H2O -NaOH-> Ar-CO-ONa + NaN3 + H2O
Alk-CO-N3 + H2O -NaOH-> Alk-CO-ONa + NaN3 + H2O
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Looking at isonicotinic acid hydrazide the the structure is more correctly

https://dailymed.nlm.nih.gov/dailymed/image.cfm?id=156505&am...
a synthetic path from the organic acid methyl ester reaction with hydrazine is shown here
http://intranet.tdmu.edu.te.ua/data/kafedra/internal/pharma_...

|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
The structure is exactly the same, simply a mirror image from the horizontal plane 

And because the molecule is also of symmetry into the screen plane...the two molecules are superimposable by 180° rotation (you of course need to
switch the -CO-NH-NH2 from side by a 180° rotation of the bond to the aromatic ring).
The NH can flip on its own and because of the vicinity of the double link...
O=C-NH- <==> HO-C=N-
[Edited on 1-6-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
It was the side chain Carbon I was looking at as the difference but it could just be the change in structure for new nomenclature where the missing C
is "understood" as I have seen that for other compounds that show the "old school" structure delineated clearly versus the "new math" diagrams that I
dislike intensely because of the ambiguous "assumed" C in the side chain 
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by Rosco Bodine | It was the side chain Carbon I was looking at as the difference but it could just be the change in structure for new nomenclature where the missing C
is "understood" as I have seen that for other compounds that show the "old school" structure delineated clearly versus the "new math" diagrams that I
dislike intensely because of the ambiguous "assumed" C in the side chain
|
I understand now .
Anyway, the second molecule you gave has stil discrete implicite C atoms for the azaaromatic ring...thus lacking 5 C and 4 H ... so practically even
if disliked...we commonly use and abuse
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Yeah there is a C in the side chain for the new nomenclature structure diagram but normal people can't see it because it is depicted in ninja
invisibility mode... so that only a special clairvoyant skilled in the ninja invisibility art will recognize it.
Obviously the "old school" classical structure diagram is more clear and more correct, and evidently the international nomenclature and standards
committee are a bunch of out of touch and "innovative" academics with their heads stuck up their asses
And then there's the whole NMR structure library discrepancy versus classical structure proofs for DDNR like Klapotke's group identified. We have an
unlikely diazo rearrangement there but nobody seems to be looking at the paradox or anomaly. Just let the computer tell you what it is .....or maybe
not! Maybe somebody should look at that issue huh? Just maybe, ya think.
The old timers just get no respect, bumping along with their old seeing eye dog who can't see either .....looking to find real love
https://www.youtube.com/watch?v=-pwC-NVPc58
<object width=640 height=480><param name="movie"
value="http://www.youtube.com/v/-pwC-NVPc58?version=3&autoplay=0&showinfo=1&modestbranding=1&controls=1&theme=dark&vq=hd720&am
p;hl=en_US&rel=0"></param><param name="allowFullScreen" value="true"></param><param name="allowscriptaccess"
value="always"></param><embed
src="http://www.youtube.com/v/-pwC-NVPc58?version=3&autoplay=0&showinfo=1&modestbranding=1&controls=1&theme=dark&vq=hd720&
hl=en_US&rel=0" type="application/x-shockwave-flash" width=640 height=480 allowscriptaccess="always"
allowfullscreen="true"></embed></object>
[Edited on 6/2/2016 by Rosco Bodine]
|
|
|
dangerous amateur
Hazard to Others
Posts: 148
Registered: 8-7-2011
Member Is Offline
Mood: No Mood
|
|
I'd like to ask another question concerning lead azide.
A description of preparation procedure mentiones growing the lead azide crystals out of a dextrine containing solution.
Afterwards the lead azide is put into a concentrated dextrine solution, filtered an dried.
The lead azide is obviously covered with dextrine on the outside, while in the first step dextrine is enclosed in the azide crystals.
What is the exact use of this second step? A phlegmatising one?
When people talk of dextrinated lead azide, what does that mean, that dextrine was used during the crystal growth, or that dextrine was used
afterwards?
|
|
|
Thraxx
Hazard to Self
Posts: 71
Registered: 15-10-2016
Member Is Offline
Mood: No Mood
|
|
My azide synthesis experiments:
Exp.Nr.1Azide Na synt.
- Bottle One :
Hydrazine sulfate 30g + 50 ml of IP alc.//+ 9,2g NaOH/-20 min. stirring
+ 9,2g NaOH,stirring 15 min,…decantacion into Bottle Two
+ 20 ml IPalc.,stirring,decantation
Bottle Two: Add to decant.sol of H.hydrate + Ipnitrite 28 ml + 9,2g NaOH prills and let stay for 40 h./The NaOH prills stood
undissolved ./
Dilute with cold ethanol ,filtration-problem with voluminous greasy soap,filtered through cloth,after diluted in 100 ml watter
,filtered with problems with the soap and precipitated with 500 ml etanol.There was ,after one day in fridge sufficiant amount of azid crystals.
Exp.nr 2-Azide K synt.- What I wanted ,and what I really did.
Prepare one big beaker 500 ml like (Bottle One) one closable bottle of 500 ml volume (like Bottle Two),one help bottle of 200 ml vol. And two cups
for KOH prills.
Prepare ice bath for Bottle One
1 Step: Into help beaker prepare 22 g of KOH diluted in 100 ml of IP alcohol.((unsuccesfully,after 24 h was not dissolved,therefore I prepared
dilution of 22 g KOH in ethylalcohol of technical grade.It was yellow-brown thick opaque dilution after 3 h.))
2 Step: Into Bottle One: 50g Hydr.sulf. (0,384 mol) + 90 ml Ipalkohol + 22 g KOH prills for 20 min stirring (( because I used ethanol in
first step,I used ethanol in the second step to.And this was mistake,because there built from hydr.sulphate not the thick paste,it was thin slurry ))
3.Step: add + 22 g KOH for second 20 min stirring and waite ====== first decantation into bottle Two
((After addition of second 22 g KOH the slurry even though after two hours decantated sharp,but the sodium sulfate was powder and allowed not to pour
the extract of.I thought,that its because watter and therefore I gave there 50 ml of IP alcohol,but it wasnt better and I must use the vacuum .))
4.Step: add + 100 ml IP alcohol - KOH solution from the help beaker ,stirring and wait for 30 min.=====second decantation into Bottle Two.
(( this was not possible,because instead of paste I had slurry which could be only filtered))
5.Step: ad + 35 ml IP alcohol stirring and decant==== into Bottle Two .
(( impossible))
6. Step: Into Bottle Two add 47,6 ml of Ipnitrite .
(( this step was well done and result was nice orange solution,where was after 20 h.in ice bath something formed on the bottom.I hope that not a
soap))
[Edited on 15-10-2016 by Thraxx]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by dangerous amateur | I'd like to ask another question concerning lead azide.
A description of preparation procedure mentiones growing the lead azide crystals out of a dextrine containing solution.
Afterwards the lead azide is put into a concentrated dextrine solution, filtered an dried.
The lead azide is obviously covered with dextrine on the outside, while in the first step dextrine is enclosed in the azide crystals.
What is the exact use of this second step? A phlegmatising one?
When people talk of dextrinated lead azide, what does that mean, that dextrine was used during the crystal growth, or that dextrine was used
afterwards?
|
Dextrine treatment afterwards (without prealable dextrin mediated cristallization) would be of no benefit since the main risk is an increase of inner
stress of large cristals (the risk of cristal abnormality is bigger into larger cristals); then the cristals may detonate under the slightest stimulus
or spontaneously.
The initial treatment allows for tiny regular cristals and that is the most important for flegmatization/phlegmatisation.
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by Thraxx | My azide synthesis experiments:
Exp.Nr.1Azide Na synt.
- Bottle One :
Hydrazine sulfate 30g + 50 ml of IP alc.//+ 9,2g NaOH/-20 min. stirring
+ 9,2g NaOH,stirring 15 min,…decantacion into Bottle Two
+ 20 ml IPalc.,stirring,decantation
Bottle Two: Add to decant.sol of H.hydrate + Ipnitrite 28 ml + 9,2g NaOH prills and let stay for 40 h./The NaOH prills stood
undissolved ./
Dilute with cold ethanol ,filtration-problem with voluminous greasy soap,filtered through cloth,after diluted in 100 ml watter
,filtered with problems with the soap and precipitated with 500 ml etanol.There was ,after one day in fridge sufficiant amount of azid crystals.
Exp.nr 2-Azide K synt.- What I wanted ,and what I really did.
Prepare one big beaker 500 ml like (Bottle One) one closable bottle of 500 ml volume (like Bottle Two),one help bottle of 200 ml vol. And two cups
for KOH prills.
Prepare ice bath for Bottle One
1 Step: Into help beaker prepare 22 g of KOH diluted in 100 ml of IP alcohol.((unsuccesfully,after 24 h was not dissolved,therefore I prepared
dilution of 22 g KOH in ethylalcohol of technical grade.It was yellow-brown thick opaque dilution after 3 h.))
2 Step: Into Bottle One: 50g Hydr.sulf. (0,384 mol) + 90 ml Ipalkohol + 22 g KOH prills for 20 min stirring (( because I used ethanol in
first step,I used ethanol in the second step to.And this was mistake,because there built from hydr.sulphate not the thick paste,it was thin slurry ))
3.Step: add + 22 g KOH for second 20 min stirring and waite ====== first decantation into bottle Two
((After addition of second 22 g KOH the slurry even though after two hours decantated sharp,but the sodium sulfate was powder and allowed not to pour
the extract of.I thought,that its because watter and therefore I gave there 50 ml of IP alcohol,but it wasnt better and I must use the vacuum .))
4.Step: add + 100 ml IP alcohol - KOH solution from the help beaker ,stirring and wait for 30 min.=====second decantation into Bottle Two.
(( this was not possible,because instead of paste I had slurry which could be only filtered))
5.Step: ad + 35 ml IP alcohol stirring and decant==== into Bottle Two .
(( impossible))
6. Step: Into Bottle Two add 47,6 ml of Ipnitrite .
(( this step was well done and result was nice orange solution,where was after 20 h.in ice bath something formed on the bottom.I hope that not a
soap))
[Edited on 15-10-2016 by Thraxx] |
You play with too big amounts and that is unsafe...you will injure yourself sooner or later.
Nitrite esters are volatile especially the nitrite ester of ethanol and isopropanol; they are also cardio-vascular/blood pressure modifier like
nitrate esters...no headaches?
Best choice is n-butyl nitrite and n-pentyl nitrite...less volatile and less flamably-explosive; allows for higher temperature work and reflux.
Soap? No soap can form...soaps are usually salts of anionic organic acids (usually with a long hydrocarbon chain) like NaO3S-Alkyl/Aryl,
LiO2C-Alkyl/Aryl; or salts of cationic alkyl/aryl-amines (primary, secondary, tertiary) like chlorides, bromides, sulfates, nitrates...
[Edited on 16-12-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Thraxx
Hazard to Self
Posts: 71
Registered: 15-10-2016
Member Is Offline
Mood: No Mood
|
|
I am careful and timid.These experiments I did on balkony in military gasmask and many pairs of gloves. But despite the fact that I did all ,I am
worried about details like touching of places and spots .Cancerogenity of hydrazine is well known and remainders of it are waiting.
After 36 h at -2 to +6 C I heat it under reflux. I heated (boiled)1 h and after I pour there 100 ml ethanol for to help precipitation.Now it is
orange solution and on the bottom excluded dark orange liquid layer .Tomorrow I will see what happened.
Sources for these experiments:
I. Video of azide synthesis : https://www.youtube.com/watch?v=qOSOXVSFdb0
II.Video is outgoing from the book : Leonid Lerner : Small Scale Synthesis of Laboratory Reagents with Reaction modeling.
This book ,pg. 118: „…while the solubility of hydrazine hydrate in isopropylalkohol is relatively poor….for this reason ethanol is chosen as a
suitable compromiss…while the solubility of hydrazin hydrate in ethanol is greater than 2,5 mol/l….230 ml of ethanolic hydrazine hydrate solution
+ 26,6 g KOH 85% + 39 g Isopropylnitrate…“
III. Sciencemadness and EWF Synthesis
1) Rosco Bodine (2004)
the hydrazine hydrate freebase which may be obtained from the hydrazine sulfate
by reaction of the solid with solid sodium hydroxide , and the absolute minimum
of added water to form a thick slurry of sodium sulfate crystals in hydrazine hydrate ,
which is taken up in successive portions of methanol . Proper technique and glassware
is needed for reactions involving the freebase hydrazine since it is destroyed
fairly rapidly by exposure to oxygen of the air .
The reaction of a slight excess of isopropyl nitrite with a cold methanol solution
of hydrazine hydrate and slight excess of sodium hydroxide , in a slightly pressurized
reaction flask produces a solid precipitate of pure sodium azide crystals in pretty good yield .
A pressure relieved , sealed glass and teflon reaction flask and addition funnel ,
with magnetic stirrer and an ice water bath is needed for performing the synthesis .
2) Microtec: 2004:
For sodium azide production I use a method which gives hydrazine in alcohol ( from hydrazine sulfate ) with little or no water:
- 1 mol dry HS is placed in a flask along with a suitable amount of anhydrous isopropanol.
- 1 mol of NaOH pellets are added and the contents are triturated with a glass rod until they begin to react, forming a slurry of hydrazine hydrate
and NaHSO4. This doesn't mix with the iPrOH, but forms a sticky goo on the glass.
- Another 1 mol NaOH is added which converts the NaHSO4 to Na2SO4 ( and forms a mol of water ) which separates cleanly as a white powder.
I would think that the Na2SO4 is a good enough dessicant to dry out the solution which can then be decanted.
- Another extraction or two with dry iPrOH recovers most of the hydrazine.
This alcoholic solution of hydrazine works well for producing sodium azide with isopropyl nitrite
__________________________________________________
Chemicals:
--------------------------------
NaOH :: mol= 39,99 m.p. =318 C,solubility – watter,glycerol,alkohol ethyl 13,9g/100ml/0C,methyl 23,8g/100ml/0C
KOH :: mol=56,105,m.p. =406 solubility-
--------------------------------
Hydrazine hydrate-:: mol=50,liquid mp= -40mboil.p.=118,5C
Soluble in :watter ,alcohol
solubility in isopropyl alkohol is poor,in ethanol 2,5mol /l
Insoluble in :eter ,chloroform
--------------------------------
Hydrazine sulphate::mol=130,12 solid m.p. 254 C,solubility at watter at 20 C =30g/l or 2,86 /100 ml
Insoluble in alcohol
---------------------------------
Sodium azide NaN3 ::
Insoluble in: aceton,ether ,chloroform,in ethanol at 0C =0,22g/100 ml
soluble in watter : 40g /100ml/10C
---------------------------------
Potassium azide KN3 ::
41g/100ml/0C , 105g/100ml/100C ,
Ethanol at 10C 0,137 g/100ml
----------------------------------
Isopropyl nitrite—density :0,87g/cm3
I)93ml (32%)HCL + 49g(61ml) IP alkohol
II) 45g NaNO2 + 80 ml H2O
Yield: 46,5g IPNitrite(53ml)
[Edited on 15-10-2016 by Thraxx]
[Edited on 15-10-2016 by Thraxx]
[Edited on 15-10-2016 by Thraxx]
[Edited on 15-10-2016 by Thraxx]
[Edited on 15-10-2016 by Thraxx]
|
|
|
Thraxx
Hazard to Self
Posts: 71
Registered: 15-10-2016
Member Is Offline
Mood: No Mood
|
|
Important is,that with the technical ethanol I had yield zero-nothing.the layer on the bottom was brown liquid in orange sollution.
Thiele s method need exact 1 mol free hydroxide for one 1 mol of hydrazine hydrate.No less,no more.If there is more,then is the reaction
going to NaNO2 and if less,then the between product ,ethylazid,stay in reaction.(I red)
Exp. Nr.1 was the case with more hydroxide.From isopropylnitrit was NaNO2 and sodium isopropylate and it was the orange "Soap"-solubile in watter and
swelling with ethanol to voluminous greasy clot.But azide was there too.
Exp.Nr2 was the same case and then the technical ethanol ,which is dilution of devil know what .
I did Experiment Nr.3 with the same rations as Nr.2,but only with IP alcohol.The reaction was slightly orange in last 6 hours of
reaction,which tooke 15 h.Yield -there was something insoluble in acetone.
I allowe me to suggest this procedure of Microtecs method:
less than 1 mol of IPnitrit /89/+extraction of 1 mol of Hydr.sulph./130/(of more than 2x 1 mol of KOH/more than 2x 56/)+ 1 mol KOH /56/(in IPalcohol
14:100) for more than 50 h.
I allowe me to tell,that the method of cold extraction publicate by Microtec is before unpublicate ,therefore original and worthy of
patent.
[Edited on 15-10-2016 by Thraxx]
[Edited on 15-10-2016 by Thraxx]
[Edited on 15-10-2016 by Thraxx]
|
|
|
TheMrbunGee
Hazard to Others
Posts: 364
Registered: 13-7-2016
Location: EU
Member Is Offline
Mood: Phosphorising
|
|
Is this really it?
So In The linked video, you can see Sodium nitrite and Urea mixed (amounts eyeballed, with slightly more urea)
Mix is then heated with direct flame,
Firstly - everything melts, some gas bubbles off (it might be even steam, CO2 and may be some ammonia) and everything solidifies,
Then it melts again, starts fizzing kind of loudly, a lot of Na ions goes in to the flame and finally explodes loudly and sharply!
It does look like Sodium azide, I could not find any real proof for this reaction, just some incomplete rumors about it. What is the equation?
https://youtu.be/VzyWsdWfGUY
[Edited on 22-12-2016 by TheMrbunGee]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by TheMrbunGee | So In The linked video, you can see Sodium nitrite and Urea mixed (amounts eyeballed, with slightly more urea)
Mix is then heated with direct flame,
Firstly - everything melts, some gas bubbles off (it might be even steam, CO2 and may be some ammonia) and everything solidifies,
Then it melts again, starts fizzing kind of loudly, a lot of Na ions goes in to the flame and finally explodes loudly and sharply!
It does look like Sodium azide, I could not find any real proof for this reaction, just some incomplete rumors about it. What is the equation?
https://youtu.be/VzyWsdWfGUY
[Edited on 22-12-2016 by TheMrbunGee] |
Nice bang! Maybe that the iron plated plate under it contributed as a catalyst...Would be nice to test this onto an inert surface like glazed ceramic.
Most probable reaction:
-No azide at all!
-Under the strong heating you get dehydration/condensation of urea into biuret, triuret and finally into melamine (triamino-sym-triazine)
H2N-CO-NH2 + H2N-CO-NH2 --> H2N-CO-NH-CO-NH2 + NH3(g)
H2N-CO-NH-CO-NH2 + H2N-CO-NH2 --> H2N-CO-NH-CO-NH-CO-NH2 + NH3(g)
H2N-CO-NH-CO-NH2 <==> H2N-C(-OH)=N-CO-NH2
3 H2N-CO-NH2 -heat-> (-N=C(NH2)-)3 + 3 H2O
Then urea, biuret, triuret and especially melamine(*) are fuels; while NaNO2 is an oxydizer.
Into the molten state both are intimately mixed at a molecular level and as such may deflagrate/detonate(**) because the average energy of all the
molecules is already high due to the preheating/melting... just like trowing an enflamed match into preheated dry wood.
Would be much more interesting to start immediately from melamine...it spares you the heating and evaporation of 3 molecules of H2O.
(*)Melamine can be considered as a trimer of cyanamide H2N-C#N.
Cyanide (and melamine) is a good way to store energy as mutliple C-N double or triple bond.
(**) A typical example is the mix of NaCN and NaNO2 what may deflagrate/detonate when molten.
[Edited on 22-12-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
| Pages:
1
..
12
13
14
15
16
..
18 |










{kind=link}
{kind=link}