| Pages:
1
..
11
12
13
14 |
jock88
National Hazard

Posts: 505
Registered: 13-12-2012
Member Is Offline
Mood: No Mood
|
|
http://www.sciencemadness.org/talk/viewthread.php?tid=30287
Lead Nitrate from Car battery + Nitric Acid
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Please don't use abbreviations without explaining them first. We're supposed to scientists, not mind readers.
I mean, do you know what ROFPM means?
[Edited on 3-5-2014 by blogfast25]
|
|
|
annaandherdad
Hazard to Others
Posts: 387
Registered: 17-9-2011
Member Is Offline
Mood: No Mood
|
|
TurboZ, if you make lead nitrate with lead and HNO3, you can get rid of the excess acid by evaporating it in a plastic desiccator with NaOH as the
desiccating agent. Using the carbonate works well, too.
Any other SF Bay chemists?
|
|
|
TurboZfreak
Harmless
Posts: 13
Registered: 11-3-2014
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by annaandherdad  | | TurboZ, if you make lead nitrate with lead and HNO3, you can get rid of the excess acid by evaporating it in a plastic desiccator with NaOH as the
desiccating agent. Using the carbonate works well, too. |
But as stated before, wouldn't that leave impurities in the lead nitrate?
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Simply recrystallize it from water, lead nitrate is freely soluble in hot water, the slight excess of nitric acid will prevent hydrolysis, and cool.
If you are patient and use more dilute hot nitric acid you can almost neutralise the acid since lead dissolves slowly even in very dilute nitric acid.
I use lead oxide or white lead pigment but the latter is is getting hard to come by these days. Lead monoxide is usually available on ebay.
|
|
|
annaandherdad
Hazard to Others
Posts: 387
Registered: 17-9-2011
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by TurboZfreak | | Quote: Originally posted by annaandherdad | | TurboZ, if you make lead nitrate with lead and HNO3, you can get rid of the excess acid by evaporating it in a plastic desiccator with NaOH as the
desiccating agent. Using the carbonate works well, too. |
But as stated before, wouldn't that leave impurities in the lead nitrate? |
No, you don't add the NaOH to the lead nitrate-nitric acid mixture, you just use it in a separate container to absorb the vapors that are evaporated
from it. HNO3 will evaporate along with the water, and NaOH will absorb both.
Or do what Boffis says, or the lead carbonate method. You can make lead carbonate by taking a sample of your lead nitrate-nitric acid mixture, and
add sodium carbonate (slowly, or else it will fizz over). This will neutralize the nitric acid and precipitate lead carbonate, which you can filter
and wash. Then add that to the rest of your lead-nitrate nitric acid mixture, to neutralize the excess acid.
Any other SF Bay chemists?
|
|
|
Motherload
Hazard to Others
Posts: 245
Registered: 12-8-2012
Location: Sewer
Member Is Offline
Mood: Shitty
|
|
I use lead metal as the limiting reagent along with 30-35% HNO3 to reduce
NO2 formation. Filter while hot to remove impurities.
After that I put the flask in the fridge to ppt as much lead nitrate out as possible.
Then I decant the mother liquor off and save it.
Then I wash the crystals in the flask with anhydrous methanol.
Pour of the methanol in a separate beaker ..... The crystals are scraped off to dry.
Within a few hours of air drying .... I have storable lead nitrate crystals.
[Edited on 6-6-2014 by Motherload]
"Chance favours the prepared mind"
"Fuck It !! We'll do it live !!"
|
|
|
woelen
Super Administrator
Posts: 8013
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
The easiest way of purifying the acidic Pb(NO3)2 is by dissolving it in as little as possible of hot water (90 C or so) and then letting it cool down.
You can allow some of the water to evaporate to regain more of your Pb(NO3)2. The crystals which separate will have lost nearly all of their acid.
Do not allow all water to evaporate. Some of the water should remain. That water will contain nearly all acid, and it also will contain other
impurities. If you allow everything to evaporate to dryness, then you'll get acid trapped in your crystals again, and you also keep other impurities.
What you can do is evaporate the water in a separate container. Then you get two batches, a large pure batch and a small impure batch.
|
|
|
jock88
National Hazard
Posts: 505
Registered: 13-12-2012
Member Is Offline
Mood: No Mood
|
|
Leas salts thread:
http://www.sciencemadness.org/talk/viewthread.php?tid=5490
|
|
|
Bert
|
Threads Merged
6-6-2014 at 06:33 |
Motherload
Hazard to Others
Posts: 245
Registered: 12-8-2012
Location: Sewer
Member Is Offline
Mood: Shitty
|
|
Previously I dried all my crystals in a glass dish .... But it's happy days are done.
So I switched to a decent quality heavy gauge Teflon coated baking tray I bought for about $10.
Anyways .... My last batch of methanol washed Lead Nitrate crystals are turning brown and the tray dark grey patches. The Teflon has not peeled off or
anything.
I am going to assume that the brown in my Lead Nitrate crystals is Iron contamination and the dark grey patches on the tray are Lead metal.
If that is true .... Then are the ions diffusing through Teflon and the crystals were just damp ?? It continued after they completely dried.
I took them off now .... I have to recrystalize the batch again.
"Chance favours the prepared mind"
"Fuck It !! We'll do it live !!"
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I discussed producing lead monoxide and lead nitrate from the lead dioxide from an old lead acid battery here:
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
| Quote: Originally posted by Hennig Brand | A while back I too took lead dioxide from an old car battery, but I didn't try and react the lead dioxide with acetic acid and I am not sure that it
would even work. I made sure the lead dioxide was well rinsed of sulfuric acid then I put it in an old steel soup can with a loose fitting steel
cover. I then put the steel can on/in a bed of hardwood coals in my wood stove and opened the draft to let more air/oxygen in and make the coals
hotter. The soup can was held at bright red/orange (ca. 650-800C) for 10-15 minutes or so at which point the can was removed and cooled. The lead
dioxide was nearly all converted to yellow lead monoxide. Yellow lead monoxide can be easily converted to lead acetate or nitrate with the
corresponding acid.
I know you said that you don't have nitric acid, but you must be able to get or make at least low concentrated nitric acid. Even 10% nitric acid can
be used, it just means removing more water to concentrate the lead nitrate solution later on. Add an excess of yellow lead monoxide to fairly warm or
hot nitric acid and then filter out what does not dissolve before concentrating the lead nitrate solution. There are a few more details, but that
should get you started.
|
I had a few pictures saved from last year that I never posted that I thought I should post here. There are pictures from a couple of different
batches. The attached instructional pdf was taken from here:
http://www.musketeer.ch/blackpowder/homemade%20lead%20acetat...
      
Attachment: Homemade Yellow Lead Oxide & Lead Acetate.pdf (358kB)
This file has been downloaded 1160 times
[Edited on 8-3-2015 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Metallic
Harmless
Posts: 2
Registered: 3-11-2017
Member Is Offline
Mood: No Mood
|
|
How is possible to remove white stain on glassware from lead nitrate? Nitric acid and aqua regia doesn't work. Maybe oxalic will help but I want to
ask before buying or synthesising it.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@Metallic, it depends what the white stains are but a good general stain remover for white lead compounds like lead carbonates halides and sulphate is
sodium thiosulphate solution, a 2-3% solution is adiquate. It sounds like your stain is lead sulphate because most other lead salts are soluble in
either acid or water. Lead halides are fairly soluble in warm water and ammonium acetate acccentuates this solubility. For lead sulphide staines treat
with 6% hydrogen peroxide for a few days until it turns white (the sulphate) and then use the thiosulphate solution.
|
|
|
Metallic
Harmless
Posts: 2
Registered: 3-11-2017
Member Is Offline
Mood: No Mood
|
|
Thank you, but I can't believe that stain is from lead sulfate because it needs to be formed from something like sulfuric acid. Actually, in another
beaker I got very similar white stain from lead acetate, but is more transparent. I will try with sodium thiosulfate but first I need to synthesis it
because I can't buy/find it in my country.
|
|
|
nezza
Hazard to Others
Posts: 324
Registered: 17-4-2011
Location: UK
Member Is Offline
Mood: phosphorescent
|
|
Here's a sample of Ammonium chloroplumbate I recently had a go at making. The procedure :-
1. Dissolve lead in acetic acid/hydrogen peroxide (Nitric acid is impossible to get now).
2. Boil down and crystallise. Dissolve the crystals in water for the Lead dioxide preparation.
3. Add excess sodium hydroxide and sodium hypochlorite to hot lead acetate solution. Mix well.
4. Filter off the precipitated Lead dioxide.
5. Add to ice cold concentrated hydrochloric acid. A yellow solution is produced (Plumbichloric acid).
6. Add saturated Ammonium chloride to precipitate out the chloroplumbate.
7. Wash with a little COLD water and dry.
Obviously wear gloves and dispose of any waste appropriately as lead is a cumulative poison.
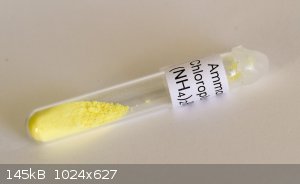
[Edited on 23-11-2017 by nezza]
If you're not part of the solution, you're part of the precipitate.
|
|
|
ninhydric1
Hazard to Others
Posts: 345
Registered: 21-4-2017
Location: Western US
Member Is Offline
Mood: Bleached
|
|
Are lead fishing sinkers pure lead or is the lead sometimes contaminated with antimony and/or other metals that help harden the lead metal?
EDIT: I happen to have a ~250 g lead sinker that I got at a sporting goods store for about $3.
(Beautiful sample of ammonium chloroplumbate by the way nezza)
[Edited on 11-23-2017 by ninhydric1]
The philosophy of one century is the common sense of the next.
|
|
|
Mabus
Wiki Master
Posts: 238
Registered: 3-11-2013
Member Is Offline
Mood: Energetic
|
|
Hard lead is almost always lead alloy, most often lead-antimony. The most pure lead is the softest.
|
|
|
teodor
National Hazard
Posts: 876
Registered: 28-6-2019
Location: Heerenveen
Member Is Offline
|
|
I prepared several lead salts using a lead-acid battery as a source of lead. May be these notes could be useful for somebody who tries to get
different pure lead salts.
The old battery I have is probably a gel battery (when I turned it upside-down no liquid was flowed out). I used a hacksaw to open the battery. I
found the best way is to cut it from a side of a negative pole. (First I tried to cut the upper cover but it led to a short circuit - you should no
try to saw the battery along the long side).

The negative plate can be easily removed. It consist of some frame and a kind of lead powder inside.

I put the plate in 480 ml of 7% HNO3. The dissolving process was accompanied by evolution of hydrogen and was slightly exotermic (it became warm but
not boiling). I covered the beaker and put it inside a bigger inverted beaker to protect the environment from toxic droplets. After few hours the
liquid became clear and the evolution of hydrogen was slowed down. pH of the liquid at this stage was around 3. I poured the liquid to a flask
(through a filtering paper), put a boiling chips and used a distillation to concentrate it to approx. 75 ml. The distilled water could be reused for
the next batch. The reason why the distillation setup should be used is a volatility of Pb(NO3)2 as well as of nitrates of some other heavy metals
which can present. I found that there is no visible salt evaporation at 50 C but in range 80-100 C volatile water-soluble salts covered the cooler
walls.
 
After cooling I added 30 ml of 53% HNO3. Pb(NO3)2 solubility drops very fast with increasing of HNO3 concentration, so this addition of the acid leads
to a big amount of precipitate. It was filtered through a vacuum filter and washed with a nitric acid of the same concentration as a mother liqueur.
After that the salt was dried. The yield of Pb(NO3)2 was 43 g. The mother liqueur as well as a washing solution as well as distilled water was used
for the next batch preparation of Pb(NO3)2.
The Pb(NO3)2 at this stage is very impure and actually can contain nitrates of all metals used for making the negative battery plate - Cd, Bi, As etc.
So, it requires a purification. I tried 2 possible method - through PbCl2 and PbSO4. For both purposes I used the same Pb(NO3)2 solution - 10-11 g of
impure Pb(NO3)2 in 60 ml H2O. At 50C it dissolves very fast and solution was clear several days without any signs of hydrolyses (I think this is
because the salt absorbed some HNO3 which preserved that).
The first method is very simple - I've added a stoichiometric quantities of NH4Cl and got PbCl2 precipitate. I think any chloride could be used -
NaCl, KCl etc. This PbCl2 could be also used as a precursor for PbO2 (didn't try that yet but NaOCl should work). There is one drawback - to recover
HNO3 the rest should be distilled with H2SO4 (otherwise you will end with very impure KNO3/NaNO3/NH4NO3). The PbCl2 is one of few insoluble chlorides
(together with Hg and Ag), so theoretically it could be quite pure but it is well known fact that when the salt deposits as very fine crystals (in
this case - like curd) it absorbs other water soluble salts from a mother liquer which is impossible to wash out. So, for better purity you can try
the second method.
The second method is a bit more complex, uses 2 unique lead reactions for better purity and allows to prepare few other salts. Add 2 ml of
concentrated H2SO4 (95-98%) per 15 ml of Pb(NO3)2 solution (of course H2SO4 with lower concentration could be also used - just use more it). The
addition of the acid deposits insoluble PbSO4. The filtrate contained HNO3 which could be used for the next preparation of crude Pb(NO3)2 directly.
The PbSO4 could be solved in concentrated NH4CH3COO and this reaction has a very good selectivity on Pb. To prepare such solution I took 115 ml of
99,5% acetic acid and added 96 g of powdered ammonium carbonate to it. The reactions proceeds by endothermic way with self-cooling to 4C so, to make
it happen I used a microwave. Several minutes (with periodic checks) on power level 100-300 W will create a clear syrupy solution without CO2 bubbles
(don't use high power - otherwise you will get a very violent reaction as well as acid evaporation). After I've got it I diluted it to the volume of
150 ml (to add some fluidity). The density was around 1.11 g/ml.
The solution such prepared could be used for dissolving fresh precipitate of PbSO4 even at cold. Actually I used the fresh PbSO4 from 15 ml of
Pb(NO3)2 solution and 2 ml and to that I added 90 ml of NH4CH3COO solution and 30 ml of H2O and warmed it a bit on a water bath. It dissolved PbSO4
almost completely. Some turbidity was filtered out. I found that addition of a small amount of water to the filtrate could make it even more clear (if
a filter paper was unable to do their job perfectly).
This solution of PbSO4 in NH4CH3COO I used to prepare more Pb salts:
- yellow PbCrO4 (by addition of K2Cr2O7). It was fail when I tried to prepare this salt from Pb(NO3)2 or PbCl2, but PbSO4 gave it perfectly.
- PbCl2 by addition of equal volumes of water and concentrated HCl, but the yield is very poor because PbCl2 forms a complex [PbCl3]- . But it could
be used for pure PbS preparation - I decanted the clear solution from PbCl2 crystals and added orange-red Na2Sx (polysulphide) solution which was
prepared by dissolving sulphur in a hot NaOH solution, it gave me pure black PbS (it was a fail to prepare this salt from crude Pb(NO3)2 (gave a mix
of sulphides with different colour - probably due to other metals contamination), PbCl2 (gave a brown lead chlorosulphide), PbSO4 in NH4CH3COO
solution (violent reaction with evolution of gases and S deposit).
It was a fail with all my tries to oxidise PbSO4 in NH4CH3COO to PbO2. The reaction with H2O2 is quite remarkable. In acidic media it doesn't react.
In alkaline media it reacts by very unusual way. When I dropped Pb solution into KOH + H2O2 it was a brown coloration (possible PbO2) which in few
seconds oxidised H2O2 and disappeared. So possible H2O2 worked as an oxidiser on the first phase and produced PbO2 but then was reduced by PbO2
formed.
Ammonium persulfate also didn't work.
It is possible to get some PbO2 from such solution by addition of H2O2 and small amount of H2SO4 till formation of a small PbSO4 precipitate and then
changing pH to alkaline with KOH. In this case PbSO4 precipitate turns to brown (PbO2?) and the reaction of H2O2 reduction and Pb oxidation goes with
the same speed, so after 24 hours there is still some PbO2 not reduced flying up and down on oxygen bubbles.
Hope this information could be useful for the topic of lead salts preparation.
|
|
|
AJKOER
Radically Dubious
Posts: 3026
Registered: 7-5-2011
Member Is Offline
Mood: No Mood
|
|
Here is one of my old threads https://www.sciencemadness.org/whisper/viewthread.php?tid=17... displaying success for a home chemist approach relating to PbO2 creation. To quote
in part:
| Quote: Originally posted by AJKOER | Successfully created PbO2 with standard household chemicals!
After dissolving my Lead source in weak H2O2/Vinegar (forming Lead Acetate), it was necessary to create an alkaline Pb salt. I added OxyClean, which
contains Sodium Percarbonate (2Na2CO3.3H2O2) and Sodium carbonate as not every household has pure NaOH or Ca(OH)2 lying around.
I believed I saw some jelly like substance and added distilled water to the mixture to this very milky/foamy solution. After a few hours, I added an
excess of NaClO. At first no obvious signs of a reaction (but a lot more foam), but within 6 hours deposits of a brownish-red salt (PbO2) appeared and
foam was nearly gone. I was hoping for a larger yield but again my Solder is low in Lead (need better cheaper source).
I am investigating other paths to PbO2 as well as using only readily available ingredients keeping in mind the goal of oxidizing ammonia in a mildly
acidic environment (which occurs, for example, using ferrate salts at room temperature).
[Edited on 24-7-2011 by AJKOER] |
This thesis https://openscholarship.wustl.edu/cgi/viewcontent.cgi?articl... clearly notes a two step process for PbO2 creation, first an acidic attack of Pb
to form a Lead salt:
Pb + HOCl + H+ --> Pb2+ + Cl- + H2O
followed by a reaction that would benefit from the presence of base:
HOCl + Pb2+ + H2O --> PbO2 (s) + Cl- + 3H+
-------------------------
Interesting, I have thoughts/research surrounding the action of PbO2 on NH4NO3 which started with this observation presented in the referenced thesis
to quote:
‘Through energetically favorable reactions, natural organic matter and pure water may reduce Pb(IV) oxides to Pb(II) species (Reaction 1.5)’
Cited Reaction 1.5: PbO2(s) + H2O = Pb2+ + 0.5 O2 + 2OH-
Under proper conditions (pKa value of NH4+ is 9.24) :
NH4+ = H+ + NH3
So, with PbO2 presence in aqueous NH4NO3, I could expect with time:
PbO2(s) + 2 NH4NO3 (aq) = Pb(NO3)2 (aq) + 0.5 O2 + 2 NH3 + H2O
This is interesting as per Wikipedia on PbO2 (https://en.wikipedia.org/wiki/Lead_dioxide ) to quote:
“2 PbO2 + 4 HNO3 → 2 Pb(NO3)2 + 2 H2O + O2
……..
However these reactions are slow”
Interestingly, Atomistry.com http://lead.atomistry.com/lead_dioxide.html on PbO2 notes, to quote :
“Hydrogen peroxide in neutral solution is decomposed catalytically by lead dioxide, but in acid solution a quantitative reaction takes place thus:
PbO2 + H2O2 + 2HNO3 = Pb(NO3)2 + 2H2O + O2;
so that lead may be estimated by titration with hydrogen peroxide solution after oxidation to dioxide by bromine in presence of alkali.”
The change to a more vigorous reaction process suggests, as I have presented in several of my recent metal dissolution threads, a radical based path.
Caution: Use only dilute hydrogen peroxide, and avoid concentrated H2O2 which can react explosively with PbO2!
So, a reaction to investigate in neutral to acidic conditions could be:
PbO2 + H2O2 + 2 NH4NO3 ---> Pb(NO3)2 + 2H2O + O2 + 2 NH3
Also, here is a paper on the conversion employing UV/H2O2 on aqueous NH4+ to NOx- with a pH product dependent, ‘Effects of pH and H2O2 on ammonia,
nitrite, and nitrate transformations during UV254nm irradiation: Implications to nitrogen removal and analysis’, in Chemosphere, 2017 Oct,
184:1003-1011. doi: 10.1016/j.chemosphere.2017.06.078, by Wang J et al, https://www.ncbi.nlm.nih.gov/pubmed/28658735 with a target pH 10.3.
The creation of nitrate is expected with excess H2O2. So, an even more preferable reaction to check out, with work required on best pH (ranging from
acidic to neutral):
PbO2 + H2O2 + 2 NH4NO3 --UV---> Pb(NO3)2 + 2H2O + O2 + 2 NH3
where, with excess H2O2, the possible conversion of ammonia as well to nitrate.
As I have previously cited on SM, a successful reaction of NH3 with Mg(NO3)2 (from freezing a mix of aqueous MgSO4 and widely available KNO3, thereby
removing the K2SO4 hydrate) as a path to NH4NO3.
[Edited on 22-10-2019 by AJKOER]
|
|
|
Refinery
Hazard to Others
Posts: 371
Registered: 17-2-2014
Member Is Offline
Mood: Still
|
|
Do lead (sesqui)silicates dissolve or decompose by any sensible mean? My local store has only this type of lead compound available.
I would suppose that PbO SiO2 compounds are as tough as aluminosilicates, aka pretty much indestructible.
|
|
|
fusso
International Hazard
Posts: 1922
Registered: 23-6-2017
Location: 4 ∥ universes ahead of you
Member Is Offline
|
|
Who has the file? The link no longer works
[Edited on 200514 by fusso]
|
|
|
Refinery
Hazard to Others
Posts: 371
Registered: 17-2-2014
Member Is Offline
Mood: Still
|
|
All right, I found some metallic lead around.
My plan is following:
I melt the lead and cast it through mesh into water to form it into shots for max surface area.
Then I get copper sulfate and react calcium acetate with it to form copper acetate, and filter.
Then I dump the shots into copper acetate solution, and a single displacement reaction should take place, forming copper metal and lead acetate.
Should this work?
I was wondering if I should re-treat the copper with sulfuric acid, dry it and send it back because the store allows returns and it's expensive...

[Edited on 14-5-2020 by Refinery]
|
|
|
Lion850
National Hazard
Posts: 517
Registered: 7-10-2019
Location: Australia
Member Is Offline
Mood: Great
|
|
Which final lead salt do you want to get to? Some time ago I needed a soluble lead salt to get to lead iodide to do the golden rain experiment. I
reacted lead fishing sinkers with hydrochloric acid (from a pool shop) with some 6% hydrogen peroxide added to encourage the reaction. It was a bit
slow but steadily reacted. I left the lead chloride in solution and then reacted that with potassium iodide (eBay) to get lead iodide.
|
|
|
teodor
National Hazard
Posts: 876
Registered: 28-6-2019
Location: Heerenveen
Member Is Offline
|
|
Lead chloride crystals
I made some nice looking lead chloride crystals today. I already described the method of lead purification by pouring 3 layers of liquid - Pb(NO3)2
solution, water and HCl acid on the top. This method allows separate lead from other impurities and get average size of crystals for easy washing. The
problem is that it is very hard to make separate layers (without accidental mixing) especially when the surface area is big like in a beaker. Without
separate layers I get a powder and I don't like powder because it is harder to wash a powder. Reaction of Pb(NO3)2 with gaseous HCl also gives a
powder. So, the idea was to try 3 layers with different densities.
ZnCl2 (bottom) - ~ 3 g/ml (6 g ZnCl3 per 3 ml H2O)
Zn(NO3)2 (middle) - ~ 2g/ml (saturated)
Pb(NO3)2 (top) - ~ 1.5 g/ml (saturated)
By this method I've got 2 different forms of crystals - prisms and "feathers". Prisms were a result of diffusion from top to bottom and feathers -
from bottom to top.
I think it is just a nice experiment to how how this interesting salt (a quite rare insoluble chloride) is growing in slow diffusion conditions. It's
interesting to check whether PbCl2 absorbs any Zn ions, but taking very high solubility of ZnCl2 I doubt it is the case.
  
(Should I try an experiment with ZnI2 ?)
[Edited on 30-8-2020 by teodor]
|
|
|
Mush
National Hazard
Posts: 633
Registered: 27-12-2008
Member Is Offline
Mood: No Mood
|
|
Method for preparing high-purity lead acetate and nanometer lead powder from waste lead paste
CN103880630A
10% acetic acid, 10% hydrogen peroxide
https://patents.google.com/patent/CN103880630A/en
[Edited on 22-9-2020 by Mush]
|
|
|
| Pages:
1
..
11
12
13
14 |









