| Pages:
1
..
8
9
10
11
12
..
14 |
sargent1015
Hazard to Others

Posts: 315
Registered: 30-4-2012
Location: WI
Member Is Offline
Mood: Relaxed
|
|
Try this?
http://www.youtube.com/watch?v=iGTq43-2V_I
|
|
|
testimento
Hazard to Others
Posts: 351
Registered: 10-6-2013
Member Is Offline
Mood: No Mood
|
|
KMnO4 isn't available for me. I could make it, but when one needs 3-6 times the amount of that for 1 part of toluene, that's just way off scale.
How about this?
Benzoic acid is produced commercially by partial oxidation of toluene with oxygen. The process is catalyzed by cobalt or manganese naphthenates. The
process uses cheap raw materials, and proceeds in high yield.
Should it need pressure, temperature? Max pressure I can reach is maybe 40-50bars with refrig. compressor. Could normal Cu salts catalyst work, or
what is the role of napthenate in here?
EDIT
This looks most promising:
https://www.google.com/patents/US3210416
Essentially one will need a steel reactor pot where toluene is inserted with cobalt or manganese catalyst and possibly a promoter, and then the vessel
is pressurized to 15-20bar and heated to 175-200C. Probably it should be mixed by jerking the pot around. This should be quite possible to produce
with common fridge pump with one-way valve and copper pipes, a needle-relief valve and a pressure meter and thermoprobe on top.
But how does this differ from Dow Process of making phenol from toluene by oxidation (Toluene + 2 O2 = phenol etc.)?
[Edited on 7-2-2014 by testimento]
|
|
|
sargent1015
Hazard to Others
Posts: 315
Registered: 30-4-2012
Location: WI
Member Is Offline
Mood: Relaxed
|
|
Pressurizing vessels at home seems... Bad? Anyways, don't blow a hand off
Also, KMnO4:
2 pounds for $20
-Sarge
|
|
|
testimento
Hazard to Others
Posts: 351
Registered: 10-6-2013
Member Is Offline
Mood: No Mood
|
|
I have more experience in that area than in chemistry.  Permanganates are banned
in my country. Permanganates are banned
in my country.
I read the patent now fully and it states much of the following:
Of total mass of toluene:
1 % manganese acetate
0.5 % ammonium bromide (i've got only sodium)
10-14bar pressure
140-180C temperature
Air bubbled inside
With high flow air it should form benzoic acid quite fast. In patent it is reported to produce several tons per hour with the specs mentioned. I think
I could use ordinary compressor because the pressure range is from 4 bars to 20 bars. 8-10 bars should be pretty enough and since the reaction goes
very fast, I can directly adjust the air flow with needle valve to compensate the consumed air(which should be mostly lack of oxygen).
Heating should be concerned, since the device is essentially a pressurized bomb which contains both oxidizer and fuel and it might go up pretty nice
if it were to get fire. So electric heating should be preferred instead of gas burner, and the exhaust gas, which certainly contains vapors of toluene
and other gunk, must be led into water trap or vented safely elsewhere or ignition may occur.
Attached picture for reactor.
The achieved benzoic acid could be turned into benzene or decarboxylated into phenol. What else could benzoic acid be used into?
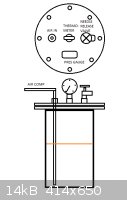
[Edited on 8-2-2014 by testimento]
|
|
|
plante1999
International Hazard
Posts: 1936
Registered: 27-12-2010
Member Is Offline
Mood: Mad as a hatter
|
|
I just had the opportunity to get 80 ml of toluene by distilling the non-aqueous layer of an old varnish can. The first test I wanted to do is
catalytic oxidation. It was just a first try using this principle.
I mixed 0.1 ml of bromine, a pinch of cobalt carbonate, and 30 ml of glacial acetic acid, I then added 20 ml of toluene and put the mixture in a 100
ml two neck flask. After the bromine addition the mixture was of a orange color. One neck was used to pass air using an aquarium pump in the mixture,
and the other had two condenser inline setup for reflux. Heating was started and the mixture slowly turned dark red, up to a point it suddenly shifted
to pink and air was passed in when the mixture suddenly changed color. Two condenser did not seem to be enough, as plenty of vapors could go all the
way out of the apparatus. At some point, white fog formed on the glass which I presume is sublimed benzoic acid.
I runned it for an hour, and then I stopped. I still have yet to find a way to extract the benzoic acid without diluting the acetic acid, as it is
quite hard to make/get in quantity.
I never asked for this.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Here’s an exploratory experiment on benzene preparation from sodium benzoate and sodium hydroxide dry distillation.
30 g of sodium benzoate and 9 g of sodium hydroxide were ground into a fine powder with a granite mortar and pestle and loaded into the following ad
hoc apparatus (here post reaction):

Because I’m not keen on using open flame with a highly flammable reaction product and rubber stoppers as sealant/connectors, I first tried heating
on full on my electrical hot plate. No product came over.
Medium propane Bunsen heat immediately caused the reagent mix to char and orange distillate to form. I reckon this needs at least 400 C to get benzene
to form. Also some white smoke that didn’t seem to want to condense formed. I kept heating somewhat intermittently until almost no white reagent mix
could be seen. The large rubber stopper had by then also suffered badly and it was time to stop.
In all I got about 25 ml of orange product with the unmistakable smell of benzene.
Here’s a close up of the reactor, post reaction: mostly black gunge with some white material too;

For a larger scale test, a metal reactor or expendable glass reactor, will be needed. I think some of the above commenters are right about heat
transfer: it appears difficult to heat the whole mass through.
[Edited on 26-4-2014 by blogfast25]
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
I'd specifically recommend Pyrex 4620/4680 dime a dozen flask auctions, and not exposing rubber stoppers to benzene, but this like everything else
will just be ignored in 3 months if not now, so there's not much point to saying anything any more.
Charring = too hot, organic chemistry = low and slow.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
S.C.:
The glass I used was cheap and I think it will be recoverable.
Rubber stoppers: agreed but this wasn't an occasion where ground glass was available to me. I'll be using another type of sealant next time, possibly
EPDM rubber with a much higher temperature/aromatics resistance.
Slow may be best but at an estimated 250 C (max setting on electrical plate) I got nothing whatsoever.
I think this preparation calls for a long, tubular type reactor geometry, placed on its long side and with gradual heating from one side to the other.
This would also prevent thermally overloading the sealant.
[Edited on 27-4-2014 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Magpie  | Benzene making, 1st picture:
<img src="http://www.scimad.org/scipics2/Na_Benzoate_+_NaOH%20_2).JPG" width="800" />
<!-- bfesser_edit_tag -->[<a href="u2u.php?action=send&username=bfesser">bfesser</a>: reduced
image size(s); hosted image(s) at scimad.org/scipics2/]
[Edited on 12.12.13 by bfesser] |
Can anyone restore these photos?
|
|
|
plante1999
International Hazard
Posts: 1936
Registered: 27-12-2010
Member Is Offline
Mood: Mad as a hatter
|
|
I doupt so, did you re-distilled the crude benzene blogfast?
I never asked for this.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
| Quote: Originally posted by blogfast25 | | Quote: Originally posted by Magpie | Benzene making, 1st picture:
<!-- bfesser_edit_tag -->[<a href="u2u.php?action=send&username=bfesser">bfesser</a>: reduced
image size(s); hosted image(s) at scimad.org/scipics2/]
[Edited on 12.12.13 by bfesser] |
Can anyone restore these photos? |
I attempted to find these 2 photos on my hard drive but they are not there.
It appears that bfesser moved them from this thread to scipics for some reason. When I go to scipics I find that the domain is for sale. Does this
mean that we have lost all the pictures on scipics forever?
[Edited on 29-4-2014 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
@plante: not yet, as the volume is so small and this was just about 'feeling the water'.
@magpie: where is bfesser when he needs a b*ll*cking? ;-)
Any chance of simply describing your set up?
I'm now thinking of a largish (30 mm OD), tubular reactor (boroglass, a large test tube in essence, I may have found just the thing on eBay),
horizontally clamped with an EPDM rubber stopper and glass tube at the neck leading to the condenser. Charge to no more than half capacity, leaving
'head room' for the expanding charge. Gentle heating by moving a propane Bunsen flame along the tube, until crude benzene starts coming off.
I'm not keen on the many Utoob metal cans + 'some sealant' + copper tube: leakage of benzene could have very serious consequences with that set up...
[Edited on 29-4-2014 by blogfast25]
|
|
|
Organikum
resurrected
Posts: 2337
Registered: 12-10-2002
Location: Europe
Member Is Offline
Mood: frustrated
|
|
| Quote: Originally posted by blogfast25 |
.....
I'm not keen on the many Utoob metal cans + 'some sealant' + copper tube: leakage of benzene could have very serious consequences with that set up...
[Edited on 29-4-2014 by blogfast25] |
The point is that a standard thread of iron watertubing, you may know the conical one, fits snuggly into the opening of a solvent can where the
plastic was removed. Some good amount of PTFE tape gives a sure seal. Just as always whe using PTFE tape -dont use force or youdestroy more then you
seal.
I am now not going into details of your suggested nonsensical setup which is so prone to failure that it hurts only reaing it. Sorry, but its true.
If you want the last edge on saftey then fit a aspirator at the receiving vessel and run it just for a little suction. Cool receiver with ice, maybe a
small condrensor with icewater on the flask before the aspirator connection.
This will speed things up and push yields a bit what will be lost by the aspirator again, but most important: no benzene nowhere never in the air.
If thats just a contest to suggest the most byzantine ways this might be implemented as the best ways are already known and just too simple, then I
apologize.
I instead will consider to contribute in this sense myself.
regards
/ORG
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
I think that a 30mm borosilicate glass tube 1/2 filled will work just fine. Keep in mind that the decarboxylation doesn't really go until the NaOH
melts. Mp for NaOH is 318°C, so this will severely corrode the glass tube. But the tube can be reused, likely for several runs (5-10 maybe).
I don't make benzene anymore - I buy it. But if you just want to make it for education/fun I understand.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
@Magpie:
Hmm... the decarboxylation works also with Ca(OH)2 and that doesn't really melt.
The glass reactor I used was unaffected by the NaOH and after washing was 'as new'. This may be due to the emanating volatiles protecting the glass
somewhat. Glass always has one advantage: you can see through it.
I'd gladly buy the benzene if I found a seller. No luck so far.
@Organikum:
"I am now not going into details of your suggested nonsensical setup which is so prone to failure that it hurts only reaing it. Sorry, but its
true."
From your hyperbolic reaction I deduce you don't seem to understand what it is that I'm proposing. You owe it to me to explain what it is you believe
that can go wrong with this 'nonsensical setup'.
Thanks for the tips on using cans, threads and Teflon.
[Edited on 29-4-2014 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
I tested the idea of a tubular reactor today, using an old Nessler tube, OD about 30 mm and nominal capacity of 150 ml:

The charge was the same as in my first experiment: 30 g sodium benzoate and 9 g sodium hydroxide, ground up (about 50 ml), spread out over the length
of the tube to allow easy heat transfer (only a thin layer of charge) and avoiding excessive expansion of the reagent mix.
I wanted to use a specially ordered black EPDM rubber stopper (better heat resistance) but it hadn’t arrived yet. So instead I used the usual (SBR?)
orange stopper but this time wrapped carefully in Teflon gas tape. There’s several cm distance between the right hand side of the charge and the
stopper, to try and keep the stopper as cool as possible.
I heated very carefully, constantly moving a medium propane Bunsen flame from one side of the charge to the other and back. But to get actual
distillate (as well as white smoke also reported by Magpie) ultimately more heat was needed. I collected about 12 g of orange distillate, a yield
based on benzene of 74 %. It doesn't appear to be possible to obtain benzene without charring the charge, at least not within reasonable time frames.
Here’s the tube after reaction:

Black patches, white patches and orange condensate on the top side.
I need to check the state of the rubber stopper to see if the Teflon wrap provided any protection.
The Nessler tube will be cleaned by soaking the inside in 30 % NaOH solution.
[Edited on 3-5-2014 by blogfast25]
|
|
|
HgDinis25
Hazard to Others
Posts: 439
Registered: 14-3-2014
Location: Portugal
Member Is Offline
Mood: Who drank my mercury?
|
|
http://web.anl.gov/PCS/acsfuel/preprint%20archive/Files/48_2...
The paper above states that, at 450º C, Sodium Benzoate may decompose to form a variety of producrs, including Triphenylmethane, wich is used to make
a large number of dyes, the Triarylmethane dyes (http://en.wikipedia.org/wiki/Triarylmethane_dye).
This is a long shot, but is there any possibility that the Triphenylmethane is reacting with other products, thus making an orange dye? Anyway, most
Triarylmethane dyes can be used as pH indicators because they change colour depending on the pH. Perhaps you could try to increase and lower the pH of
your product to see if it changes colour?
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
HgDinis25:
Quite a bit of work has gone into identifying the orange material, higher up in this thread. I can't remember the outcome, sorry. Thanks for the
paper.
I'll take you up on the pH/colour idea but my hopes aren't high.
On a more mundane level, my Teflon wrapped stopper hadn't sustained any damage worth mentioning. That worked even better than hoped for.
[Edited on 3-5-2014 by blogfast25]
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
| Quote: Originally posted by blogfast25 | | It doesn't appear to be possible to obtain benzene without charring the charge, at least not within reasonable time frames. |
Perhaps you're doing it wrong. For me there was a colorless distillate and no charring, until the heat was turned up when things tapered off, which
did nothing worth doing and should not have been done.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by S.C. Wack |
Perhaps you're doing it wrong. For me there was a colorless distillate and no charring, until the heat was turned up when things tapered off, which
did nothing worth doing and should not have been done. |
I didn't see your contribution in this thread, point to it if you will?
The first drops came over clear and colourless but at an interminably slow rate. Even at higher rate it still took about 15 min for 12 ml. At that
rate one is inclined to go full throttle at the expense of purity.
If I'm doing it wrong everyone else here, bar you (no sarcasm intended), seems to be too.
It would help greatly if someone could establish some rate-temperature relation for this reaction(s).
[Edited on 4-5-2014 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
I also had a quick stab at the reduction of phenol with zinc powder. Using the same set up as above but slightly tilted so that the Nessler’s bottom
was pointing downwards a bit. 10 g phenol and 8 g ultrafine Zn powder were then loaded into the Nessler tube.
I assume this reaction is supposed to go as:
Phenol + Zn === > Benzene + ZnO
Edit: hydrogen deleted as per deltaH (ta!)
I heated and got bubbles but no benzene. At higher heat the phenol started to distil over and froze in the condenser:

The zinc appeared also unaffected, still a grey dark powder.
It appears this reduction needs leading vapours of phenol over 400 C zinc granules or turnings, as mentioned in Wikipedia.
[Edited on 4-5-2014 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Oooopsie. You're right of course.
[Edited on 4-5-2014 by blogfast25]
|
|
|
plante1999
International Hazard
Posts: 1936
Registered: 27-12-2010
Member Is Offline
Mood: Mad as a hatter
|
|
You may want to try to pass phenol vapors over an heated tube containing zinc dust which would be heated using a bunzen.
I never asked for this.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by plante1999 | | You may want to try to pass phenol vapors over an heated tube containing zinc dust which would be heated using a bunzen. |
Sure, but it's a lot less practical as a lab preparation than a dry distillation: it requires quite a bit of engineering to do it safely, like
condensing very hot benzene for instance. The latter would require a copper coil, I think. Any unreacted phenol would have to be dealt with too, in a
separate cold trap.
Later on I'll have a quick test tube stab with aluminium powder.
[Edited on 5-5-2014 by blogfast25]
|
|
|
deltaH
Dangerous source of unreferenced speculation
Posts: 1663
Registered: 30-9-2013
Location: South Africa
Member Is Offline
Mood: Heavily protonated
|
|
blogfast, can you please ellaborate how you expect these reductions to proceed as I have a major concern:
Phenol is weakly acidic, so a reaction between phenol and an electropositive metal would most likely initially yield the metal phenate (presumable a
solid) and produce hydrogen and heat, for example:
2Ar-OH(l) + Zn(s) => Zn(O-Ar)2(s) + H2(g) + heat
Idealy, one would reflux this to prevent phenol boiling off.
The problem is that further pyrolysis of the phenate is unlikely to yield benzene because you're now short the hydrogen having previously driven it
off.
You might make diphenyl ether on pyrolysis, which would, nevertheless, be an interesting result 
?? Zn(O-Ar)2(s) + heat => Ar-O-Ar(g) + ZnO(s) ??
Note that diphenyl ether is very high boiling (258°C), so it may condense near the pyrolysis heating zone.
Apparently it smells weakly of geraniums, though I doubt a pyrolytic product would smell very pleasant 
****
I found this obscure and dated text about pyrolysing calcium phenates (not sure if phenolate or phenate is correct, the authors referred to it as
phenate, so I'll stick to that). According to them, phenol yields an 'unusual' calcium phenate hydroxide 'half salt' upon reaction with calcium
hydroxide which simply regenerates the phenol upon pyrolysis and forms CaO as co-product.
They also say that the pyrolysis of alkali phenates like sodium or potassium phenate yields only char and gas, but no volatile organic products (as
per the finding of others, see references in article). That could be on account of the high basicity of those salts, so maybe some hope for zinc and
aluminium to make something else?
See: https://web.anl.gov/PCS/acsfuel/preprint%20archive/Files/28_...
[Edited on 5-5-2014 by deltaH]
|
|
|
| Pages:
1
..
8
9
10
11
12
..
14 |









