mc68k
Harmless

Posts: 24
Registered: 26-10-2023
Location: Durham, NC, USA
Member Is Offline
|
|
Condensation of Aldehydes with Hydantoin
In another thread [1] I'm working on building a "chemputer", an automated synthesis platform. In order to test the chemputer I need a synthesis which
only requires the "modules" I've already built. So far this means liquid handling and mixing with temperature control.
This thread will record the synthesis experiments and outcomes for these test reactions to avoid diluting the hardware construction thread.
An Interesting Synthesis
Awhile back I ran into a thread where clearly_not_atara made a comment that piqued my interest:
Quote: Originally posted by clearly_not_atara  | | If the goal is homologation of benzaldehydes to one-carbon-longer amines, nitromethane can be "replaced" by hydantoin, produced by various methods.
|
I had not heard of hydantoin, so I did some research about the stated mechanism. Turns out it is quite useful!
Aldehydes will condense with hydantoin at the 5-position which can then be hydrolyzed, breaking the ring into an amino-acid, effectively adding C-N to
the original aldehyde.
I dug around SM to see if anyone had posted an experimental for this procedure, but nothing turned up. Seems like a useful technique for the amateur
chemist!
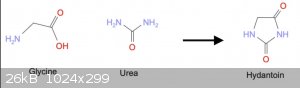
STEP 1: Hydantoin
A thread [3] mentions patent GB991644 where Glycine + Urea are fused together to give hydantoic acid, which is treated with aqueous acid to close the
ring.
Another thread [4] discusses Wada's method for making amines from amino acids (a debunked procedure, see [6]). However, the first step is to make a
hydantoin by heating Glycine + Urea in alkaline water at reflux for some time.
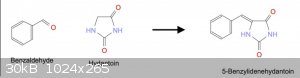
STEP 2: Aldehyde Condensation
Referring back to [2] quoted above, clearly_not_atara attached a paper (Henze, 1940) which described the condensation procedure. Unfortunately it
requires acetic anhydride, not easily acquired for the amateur (in the US).
I also found US2861079 [5] which describes a slightly simpler technique. Aldehyde + Hydantoin are combined in water with ethanolamine, heated to
reflux for some time then acidified.
Several common aldehydes came to mind to use for the procedure: Benzaldehyde, Vanillin, Anisaldehyde. For example, the condensation of Benzaldehyde
with Hydantoin would produce 5-Benzylidenehydantoin.
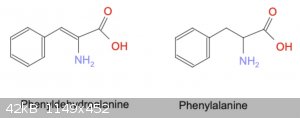
STEP 3: Hydrolysis of Hydantoin
Finally, hydrolysis of the condensation product produces an amino acid attached to the original aldehyde, effectively lengthening the carbon chain and
adding a nitrogen.
The Henze paper referred to in [2] discusses alkaline hydrolysis. Unfortunately this method requires a reduction step to eliminate the double-bond
prior to the hydrolysis. Apparently this step is necessary because the alkaline hydrolysis will split the molecule at the double bond, leaving only a
methyl group instead of the desired amino acid.
The rebuttal of Wada's method (attached in [6]) discusses acid hydrolysis conditions, which do not require a prior reduction. Interestingly, the
reduction used by Henze is to boil the hydantoin in concentrated acid - why doesn't acid hydrolysis also perform the reduction?
For example, acid hydrolysis of 5-Benzylidenehydantoin would produce Phenyldehydroalanine (phenylalanine, but with a double-bond from the benzene
ring), while reduction followed by alkaline hydrolysis would produce Phenylalanine.
Whew.. ok let's try this!
[1] https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
[2] https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
[3] https://www.sciencemadness.org/whisper/viewthread.php?tid=13...
[4] https://www.sciencemadness.org/whisper/viewthread.php?tid=99...
[5] USPTO 2,861,079
[6] https://www.sciencemadness.org/whisper/viewthread.php?tid=18...
move.w %0x2700,sr
movem.l d0-d7/a0-a6,-(a7)
lea science,a0
jsr madness
|
|
|
mc68k
Harmless
Posts: 24
Registered: 26-10-2023
Location: Durham, NC, USA
Member Is Offline
|
|
Experimental:
Glycine + Urea -> Hydantoin:
----------------------------
1.00 g (13.32 mmol) Glycine
0.85 g (14.00 mmol) Urea

Glycine and Urea were placed in a 10 mL beaker.
Approximately 6 mL water added and stirred until everything dissolved.
Heated in microwave (on low) for 1 minute, then stir.
Heating/stirring repeated for 10 minutes, never allowing the beaker to boil.
Beaker left overnight to cool and complete reaction.
Acidify to pH 2 by drop-wise addition of aqueous HCl 37%.
Acidified mixture poured into evaporation dish and warmed on hotplate.
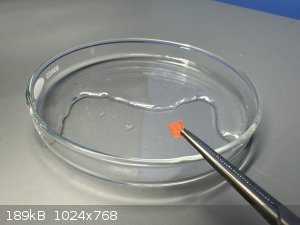
After crystals formed spontaneously the dish was left to evaporate at room temperature for 24 hours.
Crystals (still wet) were scraped together and 2 mL fresh water added to the dish, stirring the wet crystals into the fresh water to wash them.
Crystals were scraped to one side of the dish and the liquid left to evaporate again.

A day later, the crystals had not dried and the plate had gained some weight. Probably absorbed moisture from the air?
Plate was heated again and kept under heat until its weight stabilized (2.136 g).
On cooling, the crystals again absorbed water.
Discussion:
In order to check my understanding of the reaction, I did some atom counting to see what should be there, and if the extra weight is logically
accounted for by the HCl added at the end.
| Code: | Before reaction:
C2 O2 N H5 Glicine
+ C O N2 H4 Urea
= C3 O3 N3 H9 (total 1.85 g)
After reaction:
C3 O2 N2 H4 Hydantoin
+ O H2 Water
+ N H3 Ammonia + HCl => Ammonium Chloride?
= C3 O3 N3 H9 (total 2.136 g) |
I did not record how much acid was added (a few mL?), but I do have the final weight of the heat-dried mixture.
2.136 g - 1.85 g = 0.286 extra
I expect the remaining weight should be entirely HCl sequestered as Ammonium Chloride (NH4Cl), suggesting that I added 7.8 mmol of HCl to acidify the
mixture.
Given that I started with ~13 mmol of reactants, and the equations suggest that there should be 1 HCl for each Ammonia ion, then I'm at a loss as to
what's in this mixture.
So.. not sure if this synthesis worked, none of the methods describe wet crystals, they all evaporate to dryness. I'm looking for a colorimetric
method to check for carboxylic acids to see if the reaction went to completion.
One potential issue is that I forgot to adjust the pH of the initial mixture to be slightly basic. Wada uses Barium Hydroxide, the other paper
(Burton and Hu) also use BaOH, except for one preparation where Sodium Hydroxide is used. They say the mixture "was made faintly alkaline".
I'll repeat the procedure with the corrections.
move.w %0x2700,sr
movem.l d0-d7/a0-a6,-(a7)
lea science,a0
jsr madness
|
|
|
Texium
|
Thread Moved
22-11-2023 at 18:39 |
mc68k
Harmless
Posts: 24
Registered: 26-10-2023
Location: Durham, NC, USA
Member Is Offline
|
|
STEP 1 : Attempt #2
Doubling the ingredients to simplify the workup a bit.
Glycine + Urea -> Hydantoin:
----------------------------
2.00 g (26.64 mmol) Glycine
1.70 g (28.30 mmol) Urea (slight molar excess)
- Glycine, Urea, ~10 mL water added to 50 mL RBF
- Significant cooling was observed during dissolution
- Once all materials were dissolved, pH was adjusted to ~8 by addition of NaOH
- RBF heated to reflux with magnetic stirring for 3-4 hours
- pH of mixture adjusted to ~3 by dropwise addition of HCl (~2 mL)
- Mixture poured into dish to evaporate, with mild heating from hotplate
- Plate heated until weight stabilized (4.414 g)
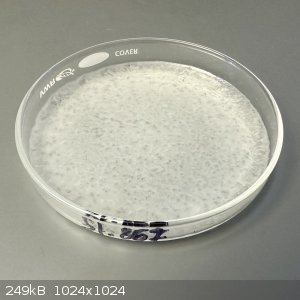
At this point I was expecting a dry powder, but the material in the dish looked like a waxy solid. On scraping, it became obvious that the material
was thick sticky sludge, like hot taffy.
To simplify the scraping process I added 2 mL water to redissolve the material. At this point, the semi-transparent waxy material turned into a
bright white flocculant, creating a thick paste. I continued to add water until all of the material had converted to the white paste.
This material is distinctly different from either of the starting materials and obviously not very soluble in water. Since Hydantoin is poorly
soluble in water (40 g/L) and the starting materials are highly soluble in water (Urea 545 g/L, and Glycine 250 g/L), this is a good indication that
the new material is indeed Hydantoin!

After thoroughly wetting and scraping the paste, the plate was evaporated with heating until its weight stabilized again. Unfortunately, the weight
just kept going down until the product began taking on some yellow color.
Presumably this was due to overheating. There's no mention of a boiling point for hydantoin on Wikipedia, so I'm guessing it decomposes around its
melting point (220 C).
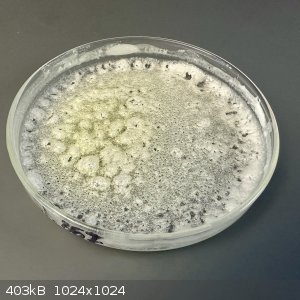
I'll discard this batch and run another experiment with lower heat.
move.w %0x2700,sr
movem.l d0-d7/a0-a6,-(a7)
lea science,a0
jsr madness
|
|
|
Loptr
International Hazard
Posts: 1348
Registered: 20-5-2014
Location: USA
Member Is Offline
Mood: Grateful
|
|
Thank you for experimenting! This is very interesting. I was on a hydantoin research kick a few years ago and recall running across some very
interesting reactions, but I can't recall them at the moment.
I just wanted to drop in and say keep it up!
"Question everything generally thought to be obvious." - Dieter Rams
|
|
|
mc68k
Harmless
Posts: 24
Registered: 26-10-2023
Location: Durham, NC, USA
Member Is Offline
|
|
Loptr, thanks for the encouragement!
STEP 1: Attempt #3 (partially successful)
- Experimental same as #2
- Observed a distinct ammonia odor from the reaction vessel (I don't recall smelling ammonia in the prior experiments)
- After refluxing for 3 hours, mixture was poured into a plate and left to dry overnight
- Crude semi-transparent crystal bursts were surround by a thick liquid
- No residual odor from the plate
- Plate weighed 5.161 g (1.461 g more than the initial ingredients)
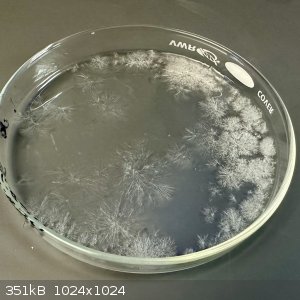
- Plate was (gently!) warmed until its weight stabilized, about 4 hours (4.452 g)
[NOTE: I'm only doing this dehydration step because the methods all distill to dryness. It is possible that water could be added to this mixture to
form hydantoin without drying it first, but I did not test that theory.]

- The crude product was scraped together, the crystals were easily collected, while the translucent areas of the plate were a resinous, sticky goop
- The methods all say this material is supposed to be Hydantoin, but it is still semi-transparent. Hydantoin should be a bright opaque white, so I'm
guessing this material is the un-closed Hydantoin, which then closes when hydrated?
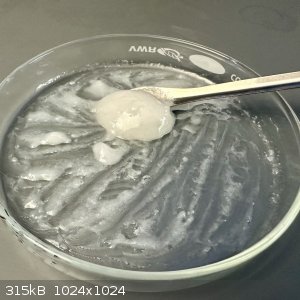
- 3 mL water was added and mixed into the material
Unfortunately, at this point the bright white paste from the previous attempt did not form. Only a light dusting of insoluble material is visible.
The rest of the goop dissolved easily in this tiny amount of water - highly likely not hydantoin.
- Additional water was added and the mixture poured into a beaker
- The beaker was left to settle (fine white crystals in suspension), about 10 minutes

- The liquid layer was poured off and the settled solids were placed on a plate to dry
- Assumed crude hydantoin solids were gently warmed on the plate until weight stabilized (1.224 g, 12.23 mmol, 46% of theoretical)
DISCUSSION:
So partial success? No characterization of the product yet.
After comparing notes from #2, the only significant differences I see are:
- Ammonia smell during reflux in #3, no notes on odor for #2
- Attempt #2 was heated two separate times (~2 h each) over the course of 24 hours, as opposed to the single 3 hour reflux in attempt #3
Likely the longer reaction time, even at room temperature, was the determining factor for success of #2.
Attempt #4 on the way...
move.w %0x2700,sr
movem.l d0-d7/a0-a6,-(a7)
lea science,a0
jsr madness
|
|
|
clearly_not_atara
International Hazard
Posts: 2786
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
1. If your product weighs more than your reactants it's dirty.
2. Acidification of the solution to pH 3 is used to precipitate hydantoic acid, not to effect a further reaction. Only after hydantoic acid is
precipitated and isolated it is added to strong hydrochloric acid and refluxed.
3. You and I must not be reading the same methods.
| Quote: | | [NOTE: I'm only doing this dehydration step because the methods all distill to dryness. It is possible that water could be added to this mixture to
form hydantoin without drying it first, but I did not test that theory.] |
15 grams of glycine, which had been freshly reprecipitated from a concentrated aqueous solution thereof by the addition
of ethanol and dried at 110 C, were throoughly mixed with 12 grams of urea, and the mixture was heated at 132 C, while stirring, until it became a
solid mass and ammonia was no longer evolved. 20 mL of water were added, and the solution was acidified to pH 3. The crude hydantoic acid which
precipitated was filtered off and recrystallized from water. Yield: 13 grams (60%); mp 173 C
comments: In this first step you are making hydantoic acid. The acid is taken up in water and dissolves by dissociating. The solution is then
acidified by another, stronger acid. This causes the hydantoic acid to become neutral (protonated) and precipitate.
21.5 grams of hydantoic acid, obtained in the manner described in the preceding paragraph, were heated under reflux for
one hour with 11 mL of concentrated hydrochloric acid and 11 mL of water. After cooling , ethanol was added and the precipitated hydantoin was
filtered off.
comments: If concentrated HCl is not available, you can use 20% HCl with no additional water. Equal volumes of conc HCl and water give roughly a 20%
HCl. Other HCl you may have can be adjusted to 20% for this step.
Anyway, I don't see anything about distilling anything to dryness here. This is just what I found in your links.
| Quote: | | - Observed a distinct ammonia odor from the reaction vessel (I don't recall smelling ammonia in the prior experiments) |
This is expected and suggests that it's working.
| Quote: | | Unfortunately, the weight just kept going down until the product began taking on some yellow color. |
To perform the recrystallization:
- take up in neutral boiling water, ideally add just enough that a clear solution is achieved at 80-100 C
- adjust to pH 3
- cool to 0 C
- filter
Only after a solid product is obtained at this step do you proceed to cyclization in strong acid. IIRC it can also be performed with ZnO but you have
HCl so that should be fine.
| Quote: | - Additional water was added and the mixture poured into a beaker
- The beaker was left to settle (fine white crystals in suspension), about 10 minutes |
This is better but you want it to be colder and with acid buffer (pH 3, I guess?) to reduce the solubility of the intermediate.
| Quote: | - The liquid layer was poured off and the settled solids were placed on a plate to dry
- Assumed crude hydantoin solids were gently warmed on the plate until weight stabilized (1.224 g, 12.23 mmol, 46% of theoretical)
|
If I'm reading the prep and your posts right, what you have is a precipitate of some hydantoic acid. Refluxing this with strong acid or with zinc
oxide should give the desired hydantoin.
But make sure that you don't use all of your intermediate on the first attempt. Start with like a third of it, so that if something goes wrong, you
can try again.
Overall, very nice, great to see some experiments with this stuff. Just some slight adjustments will make your life a lot easier, I think.
|
|
|
mc68k
Harmless
Posts: 24
Registered: 26-10-2023
Location: Durham, NC, USA
Member Is Offline
|
|
clearly_not_atara,
Thanks so much for your comments! Let me clarify the procedure I'm using so you can see where it is coming from.
| Quote: | GB991644
15 grams of glycine, which had been freshly reprecipitated from a concentrated aqueous solution thereof by the addition of ethanol and dried at 110 C,
were throughly mixed with 12 grams of urea, and the mixture was heated at 132 C, while stirring, until it became a solid mass and ammonia was no
longer evolved. |
- It is my understanding that they mean to grind the two dry ingredients together and heat the dry mixture to 132 C, only after which any water is
added?
- I was not confident that I read this correctly, so I looked for other references to corroborate..
| Quote: | Wada, 1933
1.3 g of Tryptophan, 0.5 g of urea and 0.1 g of baryte (Ba(OH)2*) were heated with 10 ccm of water. After three hours some HCl was added and
everything was evaporated to dryness in vacuum. [it is my understanding that the remaining steps describe alkaline hydrolysis] |
- This seems much simpler and is roughly the method I'm using (see adjustment by Burton & Hu)
- No characterization of the hydantoin is given
| Quote: | Burton & Hu, 1949
p-Sulphamylphenylalanin hydrochloride (2 g.) in water (20 c.c.) was made faintly alkaline with dilute sodium hydroxide, urea (1 g.) added, and the
solution refluxed for 3 hours. The cooled solution was acidified with dilute hydrochloric acid and evaporated on the steam-bath. The crystalline
hydantoin was collected, washed with a little cold water. and air dried: yield 1.4 g. (68%), m. p. 206-207" (decomp.)
Following Wada's method (Zoc. cit.), glycine (5 g.), urea (5 g.), and 0.4N barium hydroxide (25 c.c.) were refluxed for 3 hours. The solution was then
acidified with sulphuric acid, filtered, and the filtrate evaporated almost to dryness. [it is my understanding that the remaining steps describe
acid hydrolysis]
Tyrosine (decomp.325-326") (2g.),urea (1g.), and barium hydroxide octahydrate (0.2 g.) in water (100 c.c.) were refluxed for 2 hours, filtered hot,
and the filtrate acidified with hydrochloric acid. Evaporation then gave the hydantoin, m. p. 264". |
- The first preparation uses Sodium Hydroxide instead of Barium Hydroxide
- Both the p-Sulphamylphenylalanin and Tyrosine preparations specifically state that the hydantoin (with characterization) is precipitated after
reflux of the mild alkaline mixture followed by acidification and evaporation.
The ingredients I chose were:
- Water
- Urea
- Glycine (because I want to make the unsubstituted hydantoin)
- Sodium Hydroxide (I don't have Barium Hydroxide)
- Hydrochloric Acid (though I also have sulfuric acid)
And all of the water-based preparations state:
- Mix ingredients in water
- Make alkaline
- Reflux the alkaline mixture for 2-3 hours
- Acidify and evaporate to dryness (sorry - not distilled, just evaporated)
- The result is a hydantoin, with various methods to recrystallize
Your comments seem to extend the description of the GB991644 process, but I'm not sure how to apply them to the water-based method I've selected:
For example, why does the GB991644 procedure require an extra precipitation step for the hydantoic acid which the water-based preparations don't
require?
If the water-based methods are producing hydantoic acid during reflux, then why does it not precipitate when acidifying the mixture like the patent
describes?
The water-based methods don't reflux the acidified mixture (although some preparations mention evaporation via steam bath), is reflux of the acidified
mixture necessary?
Thanks!
[Edited on 27-11-2023 by mc68k]
move.w %0x2700,sr
movem.l d0-d7/a0-a6,-(a7)
lea science,a0
jsr madness
|
|
|
mc68k
Harmless
Posts: 24
Registered: 26-10-2023
Location: Durham, NC, USA
Member Is Offline
|
|
STEP 1: Attempt #4
- Same experimental as #2
- Initial pH adjusted to pH 9 (little too much NaOH)
- Held at reflux with stirring for 6 hours
- Kept better track of the odor:
- Initial solution had no discernible odor
- After 45 min reflux, the ammonia odor was just barely present
- After 2 hr reflux, the ammonia odor was quite potent
- After 4 hr reflux, the ammonia odor was still present, but not as potent
- After 6 hr reflux, ammonia odor was still present
- After acidifying the mixture, no ammonia odor was present
- Poured acidified mixture into a plate and gently heated it for awhile, then left overnight to dry
Unfortunately, like attempt #3, most of this product dissolved easily in 2 mL water. It did seem like there were more solids this time, but nothing
like the quantity from attempt #2.
As I was writing up this procedure, clearly_not_atara commented on the process. One comment caught my attention:
| Quote: | | what you have is a precipitate of some hydantoic acid. Refluxing this with strong acid or with zinc oxide should give the desired hydantoin
|
The procedures I'm using don't explicitly state that they reflux the acidified mixture, however in some cases they did use a steam bath to speed
evaporation. Meanwhile the patent explicitly states this step.
Some analysis of the papers and my attempts:
- Wada describes "evaporated to dryness in vacuum" which I assume is not high-heat
- In Burton & Hu, of the 6 hydantoin preparations 4 of them imply that they were "evaporated on the steam-bath" (one preparation explicitly,
followed by 3 more that were "similarly obtained").
- Likewise, my attempt #2 (which appeared to work) heated the evaporation plate too hot, possibly mimicking the steam bath of Burton & Hu
- My attempt #3 and #4 both use gentle heating to evaporate the mixture, and both returned a similar results, only partially successful
Since I had just finished evaporating the acidified mixture to discover it was not a success, I decided to extend the experiment slightly:
- The material was re-suspended in some water and placed in an RBF
- pH was tested to be ~3
- The mixture was heated to reflux for 1 hour (this should simulate the action of the high-heat evaporation method)
The mixture was then divided into two portions:
- Portion #1 was placed on a plate to evaporate, it had no discernible odor
- Portion #2 received an additional 2 mL conc. HCl (should be equivalent to the patent's 1:1 HCl:water environment)
- Portion #2 was heated to reflux another hour, then placed on a plate to evaporate, it had a slight odor that I did not recognize (specifically, it
did not smell like HCl)
This gives me 3 data points:
- No heat evaporation, result was the same as attempt #3
- Mild acid at reflux, result should simulate attempt #2 and Burton & Hu
- Strong acid at reflux, result should simulate the last steps of the patent
High-Heat, pH 3:

High-Heat, pH <= 1:

Looks like clearly_not_atara's suggestion is right on the money!
Now I just need to characterize this material to check the result.
move.w %0x2700,sr
movem.l d0-d7/a0-a6,-(a7)
lea science,a0
jsr madness
|
|
|
clearly_not_atara
International Hazard
Posts: 2786
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Barium hydroxide is used to bring about a variety of alkali-catalyzed reactions, some of which just don't work quite as well with anything else. I
have never really understood why. For example diacetone alcohol is always made with baryta. In this case it seems like there are condensations with no
barium though.
I think the crystals look encouraging. Not sure why I thought a reflux with acid was necessary. Hope it turns out as desired.
I'm tired so I'll type more later.
|
|
|
mc68k
Harmless
Posts: 24
Registered: 26-10-2023
Location: Durham, NC, USA
Member Is Offline
|
|
STEP 1: Attempt #6
Q: What happened to attempt #5?
A: We don't talk about attempt #5.
A bit frustrated, I decided to look for additional literature and stumbled upon a masterpiece! Patent (US4647694) which is using very similar
water-based methods and not isolating the intermediate hydantoic acid. It also has a specific example of hydantoin synthesis from Glycine and Urea
with detailed workup instructions.
| Quote: | US4647694
PROCESS FOR THE PREPARATION OF D1-B-ARYL AMINO ACIDS
According to the process of the present invention hydantoine of the Formula I is prepared with a yield of 70-75% by heating to boiling an aqueous
solution of glycine and an excess of urea (without isolating the hydantoin acid intermediate) and subjecting the mixture to acidic cyclization. At the
end of the reaction the desired product precipitates in crystalline form and can be directly used in the next step.
...
The ratio between the glycine and the urea that is preferred in the first step of the new process is a molar ratio of 1:1.8 to 1:5, preferably 1:2.3.
...
To facilitate the cyclization reaction to form the hydantoin, sulfuric acid is then added in concentrated form in an amount of 1:0.3 to 1:1,
preferably 1:0.5 relative to the glycine.
EXAMPLE 1
Hydantoine (I)
75 g (1 mole) of glycine and 140 g (2.3 moles) of urea are dissolved in 120 ml (6.7 moles) of water and the mixture is heated to boiling for 12 hours
under stirring. To the reaction mixture 90 ml (1.7 moles) of concentrated sulfuric acid are added dropwise under cooling with ice cold water and
stirring. The reaction mixture is heated to boiling for an hour and thereafter cooled to 0°-5° C. The hydantoin precipitates in crystalline form.
The crystals are filtered off, washed with cold water. A further amount of the product can be obtained by evaporating the mother liquor. Thus 71 g of
the desired compound are obtained, yield 70%, mp.: 223°-226° C.
The crude hydantoin thus obtained is sufficiently pure for further reaction. It can be recrystallized from 10 parts of glacial acetic acid, mp.:
220-222° C. |
Some differences: they are using a much larger ratio of Urea, no Sodium or Barium Hydroxide, and acidifying with Sulfuric Acid instead of HCl.
Glycine + Urea -> Hydantoin
----------------------------
Using 1/10 scale of ingredients for "US4647694, Example 1":
-------------------------
7.5 g Glycine
14 g Urea
12 mL Water
9 mL Sulfuric Acid
- Dissolve Glycine and Urea in water
- Reflux for 12 hours
- Cool mixture in ice bath (to allow safely adding Sulfuric Acid)
- Dropwise, with stirring, add conc. Sulfuric Acid
- Reflux for 1 hour
- Cool mixture to 0-5 C to precipitate the hydantoin crystals
Yield of 70-75%, mp. (crude): 223°-226° C
Recrystallize from GAA mp.: 220-222° C
Experimental:
11:15 - Mix Glycine, Urea, Water. This quantity of reactants does not readily dissolve. Much agitation was used and finally the undissolved mixture
was setup for reflux anyway
11:30 - Begin heating mixed ingredients (mixture finally dissolves upon reflux)
12:30 - No scent
13:30 - Very sharp ammonia odor
16:30 - Crystals have formed in the reflux condenser
21:30 - Only 10 hours of reflux
- Cool flask
- Add sulfuric acid dropwise
- During addition, mixture became cloudy
- Near completion, cloudy layer floated and conglomerated, a glass stick was used to break up the clot
- Acidified mixture measured to be pH 1-2
- After addition, RBF was brought to reflux again for 1 hour
- Upon heating, the clot redissolved
22:30 - Remove from heat and cool overnight
Success!

After filtering and washing, there remained beautiful fluffy white crystals!

I'm quite pleased with this reaction, it is exactly as described and required no guesswork on my part.
Still need to recrystallize, and find some method of characterization.
--------
US4647694 : PROCESS FOR THE PREPARATION OF D1-B-ARYL AMINO ACIDS
Attachment: US4647694.pdf (593kB)
This file has been downloaded 145 times
move.w %0x2700,sr
movem.l d0-d7/a0-a6,-(a7)
lea science,a0
jsr madness
|
|
|
|








