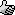SyntheticFunk
Harmless

Posts: 21
Registered: 30-12-2022
Member Is Offline
|
|
Synthesis of Nitrotoluene
Hi all. This is my first post on Sciencemadness so I apologize if I commit any errors of etiquette or of any other nature. I happily welcome any
corrections or advice.
Recently, I have had the need to make o-nitrotoluene for indole ring synthesis. I've done a brief search through relevant literature and a more
detailed search on the forums here. The procedure I followed is, in fact, nearly identical to the one posted by Magpie here: http://www.sciencemadness.org/talk/viewthread.php?tid=29111
Thank you for sharing, by the way, Magpie. It was very helpful.
I've decided to post my synthesis as corroboration of results by replicating previous experiments is an important (and often overlooked) component of
science. I've also run into some issues regarding the separation of the ortho and para isomers and was hoping to get some insight, but I'll discuss
that more later in the post. I apologize in advance for the poor formatting of photos, I am unfamiliar with how to format correctly on this forum.
Reaction:
3*C6H5-CH3 + 3*HNO3 -> o-C6H4(CH3)-NO2 + m-C6H4(CH3)-NO2 + p-C6H4(CH3)-NO2 + 3H2O
CAUTION - copy pasted from Magpie's post
Nitrotoluenes are poisonous and possibly carcinogenic. NOx vapors are poisonous. Use adequate ventilation and wear gloves.
Excessive temperatures can produce dinitrotoluene and/or trinitrotoluene, which can be explosive. Keep the temperature under 30°C during the acid
addition.
An additional word of warning from myself. Both nitric and sulfuric acid are extremely corrosive with nitric acid also being an oxidizer. Be prepared,
have an acid spill kit somewhere on hand just in case. Believe me, you never know when you'll need it... (ominous foreshadowing)
Reagents:
Toluene (42.7g)
68% Nitric Acid (29.6mL)
96% Sulfuric Acid (34.3gmL)
Part A: Preparation of the nitration mixture
To a 1000mL beaker cooled with an NaCl-ice bath was added 34.3mL of 96% sulfuric acid. A 125mL separatory funnel was filled with 29.6mL of 68% nitric
acid and the nitric acid added to the sulfuric acid dropwise over a ~30 minute period, occasionally agitating the beaker to aid mixing. White fumes
are observed upon the addition of nitric acid with a blue glove placed to aid in visualizing the fumes.
Part B: Addition of the nitration mixture to toluene
A 500mL 3-neck round bottom flask is equipped with a stir bar, a reflux condenser (to contain any splashing, no water was flowing through), a
thermometer, and an addition funnel containing the nitration mixture. To this flask is added 42.7g of toluene and it is pre chilled using a NaCl-ice
bath prior to the addition of the nitration mixture. After the toluene has sufficiently chilled (~-10C), the nitration mix is added SLOWLY with
vigorous stirring. Approximately one drop is added every 2-3 seconds and the temperature remained between -5C and 0C throughout the entirety of the
addition phase, which in total takes ~2hrs. Aluminum foil surrounded the ice bath with the intention of insulating it to minimize replenishing the
cooling bath. The reaction gradually became more red in colour as it proceeded.
Part C: Warming of the Reaction Mixture Following Completion of Addition
Once the nitration mix has been completely added the cooling bath is removed and the reaction mixture is allowed to heat on its own with a target of
45C. The reaction does, in fact, produce enough heat to allow it to warm to 45C, but is struggled to maintain this temperature, and after one hour
heat had to be supplied, though very little (setting 2-3 on the hot plate, which would likely have been too much if the round bottom had better
contact with the hot plate). The flask is maintained between 30-45C for one more hour (2 hours total) before it is removed from heating by slightly
raising it above the hot plate.
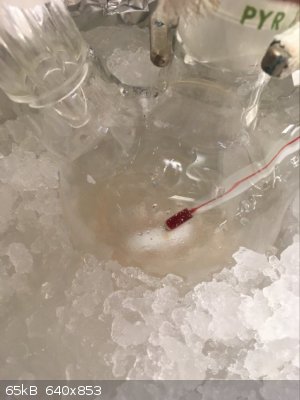
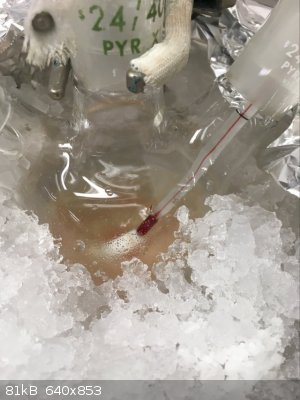
Part D: Cry because the Clamp Slips/Settles Shortly After You Raise the Flask Causing it to Smash
Aaaaaand this is where it went wrong. Our damn rusty clamps tend to feel tight as if they will not tighten anymore despite not truly being tightened
all the way. The clamp shifted a few minutes after raising the flask and despite the fall only being 3/4"-1", it was sufficient to break the flask.
Thankfully I was working in a fume hood, with PPE and a proper spill kit nearby. I would hate to have had to deal with this in any other setting. It
already sucked as is.
Spill X-A Acid Neutralizer (SDS says it containers a mixture of MgO, Fuller's Earth, and Na2CO3) is generously applied to the fume hood to prevent the
contents the reaction from spreading everywhere. Somewhere in between between the expletives I was yelling out I realized I still might be able to
recover a bit of product as nearly all of it had been captured by the absorbent. With renewed hope I carried on to the work up. From here forward I
have no more pictures, in the chaos of it all I simply forgot.
The absorbent is swept up and placed into 500mL of water. The pH is measured using indicator strips and was found to be ~10-11. Perfect. The first
step of the work up is neutralization anyways, and 500mL of water has to count as a wash right? Right??
The ungodly paste/water mixture is vacuum filter in batches, the aqueous filtrate collected. The solids in the filter are transferred to a beaker and
2x100mL of dichloromethane (DCM) is added to the Buchner funnel to try and extract any product that may be stuck in the solids. In total, 2
filtrations are required to thoroughly remove the solid mass resulting in 400mL of DCM that was collected. The aqueous layer itself is extracted
2x100mL of DCM as well, bringing the total up to 600mL. The DCM is dried with large granules of CaCl2 and the DCM/nitrotoluene mixture is simply
decanted off into a 1L round bottom flask where it is evaporated under reduced pressure using a rotary evaporator. The DCM collected for later use in
experiments where purity is not so important or I will further distill is if required. After the DCM is removed, the rotary evaporator water bath in
increased to 65C and 100mmHg (enough to remove the toluene according to the nomograph tool by Sigma-Aldrich). The crude product is an amber coloured
oil weighing 32.8g (51.6% yield). I'm quite shocked considered the bench top extraction that occurred.
Part E: Purification/Separation of the ortho/para Isomers
Based on my readings, the purification follows one of two routes, or more often a combination of the two. Purification by cooling to -20C, filtration
of the resultant p-nitrotoluene crystals, rinse repeat. Or by fractional distillation either of the crude mixture as is, or of the slightly purified
oil following the aforementioned purification by precipitating the para isomer. I've attempted the former, cooling to -20 and filtering any crystals.
This has been relatively unsuccessful. Left alone, no crystals form. However, some strong agitation causes some crystals to precipitate out, though
they are short lived and filtration has caught no crystals in the filter paper. I thought some residual toluene may be to blame, but further
rotovap'ing results in oily reside/crystalline material collecting in the bulb (not the trap, the bulb in case of bumping). Based on this I fact I
reckon I have either completely, or nearly completely removed all of the toluene.
From here I am faced with a few options. I can cool it further (I have access to liquid nitrogen and a -78C freezer) and hope for precipitation, an
appealing option though not guaranteed to work. Alternatively I could fractionally distill it, though this thought is mildly unsettling given the
potential explosion hazard. Chromatography would be a nice alternative, though I have not read of this being done. If anybody has some advice on the
separation of isomers I would be greatly appreciative. And of course I welcome any criticisms or comments on the procedure as a whole. I realize that
posting this procedure with the somewhat embarrassing feature of having the flask contents spill all over the fume hood risks making a fool of myself
on my very first post. But I think it is important to share the failures in addition to the successes as well as highlight the fact that accidents can
very easily occur. Always have a plan for if/when things go wrong, nobody ever really expects that they will. Let my failures be a shining example of
what you do not want to happen. However, given I was still able to recover a ~52% yield I hope this helps others realize that spilling the contents of
your reaction is not always the end of the world, it can sometimes be recovered.
Thanks for reading. Stay funky.
[Edited on 22-1-2023 by SyntheticFunk]
|
|
|
CouchHatter
Hazard to Others
Posts: 152
Registered: 28-10-2017
Location: Oklahoma
Member Is Offline
Mood: 76 elements taken!
|
|
Welcome, and what a great post with pics. RIP flask. Good job on the recovery. We like failure around here just fine, that's how you learn what works!
Magpie passed away in 2019, and his nitrotoluene post is indeed immaculate. Also instructive might be Urbanski Vol 1, p273-4(available on
archive.org). He discusses in depth the ideal nitration ratios of acids to toluene; what worked well for me was dropping the pre-mixed, pre-chilled
acids, and the toluene, in equal measure into the flask with good stirring and cooling. After the secondary heating and successive neutralization
washes, my freeze precipitation did yield some p-nitrotoluene crystals, so obviously here we diverge. And yet... Mine reached higher temperatures than
yours, and even formed a smidge of dinitrotoluene, which I discovered in the bottom of one of the beakers. I vacuum distilled the crude washed product
and nothing went awry.
Autoignition temp is different than flash temp. I don't want to say heating the crude is NOT dangerous, but the danger of explosion if there are no
di- and tri- compounds in your mixture from over-heating during nitration is minimal. That's how I understand it, and I never did have a problem with
my vacuum distillation and successive fractional distillation. After distilling over most of the crude, I did NOT distill to dryness, in fact I
stopped when the undistilled pot became very dark. I used a cold trap, and I only heated as much as needed to collect a slow, steady stream of
distillate. Nothing special. Then the distilled contents freeze-precipitated even more crystals, then redistill, etc.
In this way I obtained quite pure bright yellow o-nitrotoluene liquid (that does not freeze precipitate anything), as well as a lot of white
needle-like p- crystals contaminated with m-.
I set my deep freeze to -20ºC, so if you're reaching that and nothing is crystallizing, either you have a good amount more o-nitrotoluene already
(possible! depends on nitration protocols), or there's no p-nitrotoluene in there (unlikely). -78ºC might be worth a shot, I suppose, but from the
sound of all that it went through, you'd want to redistill it anyways before using it.
Here's a photo mid-stream, before fractional distillation of the final product.

|
|
|
Keras
National Hazard
Posts: 931
Registered: 20-8-2018
Location: (48, 2)
Member Is Offline
|
|
I didn't know sulphuric acid was need for mono-nitration. It is definitely needed for bi- or tri- nitration, because you have to overcome the
deactivation linked to the attachment of the first nitro group, but toluene is relatively activated w/r to vanilla benzene, by virtue of the slightly
donor methyl group.
Isn't o-nitrotoluene steam distillable whereas p-nitrotoluene is not? Steam distillation is way easier to carry out than fractional distillation, let
alone fractional crystallisation which would require all your apparatus to be cool down to negative twenty
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
SyntheticFunk - well done and welcome here!
o-nitrotoluene must be vacuum distilled through very powerful column with good packing and adjustable reflux ratio head... in my experiment I used 600
mm long 30 mm inner diameter Hempel and the purity of o-nitrotoluene was only 83%, the rest m- and p- isomers :-( and I distilled a mixture after
partial crystallization of p-isomer... maybe using 1000 mm long Hempel column could be enough... vacuum distillation using column is much less
efficient than at atmospheric pressure due to low pressure of vapor
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
Keras - steam distillable is o-nitrophenol.
[Edited on 23-1-2023 by Fery]
|
|
|
SyntheticFunk
Harmless
Posts: 21
Registered: 30-12-2022
Member Is Offline
|
|
Thank you for the warm welcome CouchHatter and Fery! I've been a long time lurker and thought it was about time I join.
Rest in peace Magpie, I'm sorry to hear that he's passed. I've come across his name often on these forums, he was a talented chemist.
Thanks for sharing a photo of the various samples of nitrotoluene CouchHatter, it gives me a rough idea of the composition of my crude sample. I would
say it is very close in colour to the sample that is third from the left side. Definitely amber in colour. I agree after a bench top extraction it
definitely needs more purification than the recrystallization procedure outlined.
I have a decent sized Vigreux column (~450mm, maybe larger) and a lot of 5mm glass beads to pack it with. In previous fractional distillations I've
had issues with flooding when I used smaller beads for packing so I'm hoping these larger ones won't be such a pain. The only issue I forsee is that
my vacuum pump is not the best and only gets down to ~10-20mmHg (bp p-nitrotoluene = 107C @ 10mmHg, ortho-nitrotoluene = 94C @ 10mmHg calculated from
Sigma-Aldrich nomograph). This is still below the explosion risk temperature I've seen mentioned at 160-170C, but of course with a fractional
distillation I'll have to heat the mixture higher than the values I calculated to force the vapours through the fractioning column.
It'll probably be fine and be well below the temperature that worries me and I'll probably end up doing this as it is the most reasonable way to
purify it. I found an interesting article on separating aromatic nitro compounds by reverse phase liquid chromatography so I may try this with a small
quantity and see if it's viable. The only C18 I have is intended for flash columns (our flash chromatograph is currently out of order), but I reckon
it'll work if I just hook it up to compressed air.
Thanks Fery for sharing the specs of your fractional distillation set up! It provides me with some realistic expectations of what I'll need to use to
accomplish this separation.
I haven't had a chance to check up on my product since the day I posted this. I'll check it later today and see if a few days in the freezer has
helped with crystallization. Will also run some TLC so see if chromatography is viable and post my results. I used to hate running columns, but they
can be run pretty fast, faster than distillations at least so I've warmed up a little.
Update:
It appears that patience will be rewarded! I checked the vial of product today and found what appeared to be quite a large mass of crystals.
A Buchner funnel is prechilled to ~-10C and then used to vacuum filter the solid/liquid mixture. The solid material weighs 3.2g, mostly white with
some yellow patches while the filtrate was weighed as 28.2g and has lightened in colour. The crystals are stored to later extract more of the Ortho
isomer that is trapped in the crystal lattice while the filtrate is returned to the -20 freezer for an additional round of precipitation.
Attachment: ReversePhaseChromatographyofAromaticNitroCompounds.pdf (295kB)
This file has been downloaded 268 times
 
[Edited on 25-1-2023 by SyntheticFunk]
|
|
|
SyntheticFunk
Harmless
Posts: 21
Registered: 30-12-2022
Member Is Offline
|
|
Quote: Originally posted by Keras  | I didn't know sulphuric acid was need for mono-nitration. It is definitely needed for bi- or tri- nitration, because you have to overcome the
deactivation linked to the attachment of the first nitro group, but toluene is relatively activated w/r to vanilla benzene, by virtue of the slightly
donor methyl group.
Isn't o-nitrotoluene steam distillable whereas p-nitrotoluene is not? Steam distillation is way easier to carry out than fractional distillation, let
alone fractional crystallisation which would require all your apparatus to be cool down to negative twenty |
Thanks for the input Keras. Whether or not sulfuric acid required is a good question. I would love the opportunity to save on reagents. I agree that
the alkyl group activation makes mononitration relatively facile compared to the dinitration or the nitration of benzene, for example. However, each
literature source I've read includes it (though admittedly without much explanation). But there are two functions I reckon that the sulfuric acid is
performing.
1. Protonation of HNO3 to drive the formation of the NO2(+) electrophile. Admittedly, this could be accomplished with excess nitric (or
possibly fuming nitric), but I am unsure plus nitric acid is more difficult to acquire for me anyways.
2. Dehydration of the water produced during NO2(+) formation. Now this may be a dubious claim. Since everything is mixed how can the sulfuric acid
dehydrated the reaction mixture? I suspect that rather than dehydrate the solution itself, it helps rip the H2O(+) off of H2O(+)-NO2. The affinity
for water by sulfuric acid would also likely prevent the reverse reaction where water formed reforms with NO2(+).
This could all well be true, but I suppose it doesn't answer the question of "is it necessary?". Possibly not, but something has to donate protons and
sulfuric is a cheap choice with potential added benefits.
As for the steam distillation. I had read about this being used for purification. I've historically had miserable luck with steam distillations so
I've had a bit of an aversion to them. But based on the GC-MS I obtained comparing the pre- and post- fractionally distilled, an alternate method is
desirable as separation was not very significant.
Regarding fractional crystallization. I am in complete agreement that it's a miserable route to go and unlikely to result in an appreciably pure final
product. However it doesn't require an entire apparatus to be cooled, really just a vial or bottle that can contain your product. The former makes it
sound more cumbersome than it really is. But overall I agree, it's not ideal.
|
|
|
|