Boffis
International Hazard

Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Mononitration of bromo and fluorobenzene
On the SM site there is an excellant account of the dinitration of bromobenzene to 2,4-dinitrobromobenzene but there is no such account of the
mononitration and the separation of the isomers. I have been working on this recently and while my experiments have been highly successful there is an
issue with the mixing. To obtain a high yield it is essential to mix the bromobenzene with the mixed acids but towards the end this becomes very
difficult and after the latest batch my arms are so stiff from 1.5 hours of constant manual stirring and then kneading the product into the mixed acid
I think they are about to fall off! Niether magnetic stirring nor an overhead stirrer is any use towards the end. Without it 10-20% of the
bromobenzene is recovered unchanged but with prolonged kneading this is reduced to zero and the yield of mixed isomers is practically quantitative.
Is there anything that can be done to improve the homogeneity of the reaction mixture? I wondered about the use of anhydrous nitric acid in DCM
discussed on SM a while back, any thoughts on this?
On a similar vein has anyone got an actual procedure for the nitration, both mono and di-, for fluorobenzene or a ref to one? I have found several
papers that discuss the theoretical aspects of this nitration which is surprisingly faster that that of either chloro or bromo benzene and the mono
nitration is heavily biased toward the p-isomer (which I am interested in).
|
|
|
njl
National Hazard
Posts: 609
Registered: 26-11-2019
Location: under the sycamore tree
Member Is Offline
Mood: ambivalent
|
|
I would definitely give the dcm nitration a shot. You might then be able to get away with less sulfuric acid and therefore have a less viscous
reaction mixture. Maybe you could even try to remove the water with a dean stark trap by refluxing with dcm? I know the azeotrope has very little
water in it but it might be worth a try.
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Hi Boffis,
I have zero experience with this reaction but this fact doesn't prevent me from thinking aloud. :-)
What if you used some sort of additive, which is unreactive towards nitration, has low viscosity, dissolves organics fairly well, has a low enough
boiling point as to not be a b.tch in the workup phase?
I mean, maybe simply diluting the reaction mixture with a few (hundred?) milliliters of DCM (or chloroform maybe) would do the trick...
I don't know the required temperature but I assume if you were kneading it in the end with your hands then it had not been too hot. (I know... I know:
you were probably using some sort of glass or porcelain rod to touch it and not your hands in a glove.)
The nitration with anhydrous nitric acid in DCM is probably a good method too but as far as I remember the solubility of nitric acid in DCM is very
limited, 1-2% comes to mind. If you were using too much acid then would end up basically in my above proposed method - without the sulfuric acid. :-)
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Pumukli, I have just completed an attempt at nitration of bromobenzene using dichloromethane as a reaction medium. The reaction was carried out
using 25g of bromobenzene in 30ml (c 40g) of DCM. I initially used sodium nitrate in sulphuric acid as the nitrating agent but discovered a few
problems and ended up adding a little 70% nitric acid to help provide water to improve the solubility of the sodium nitrate/bisulphate. When quenched
with ice the organic layer becomes butter-like and it is better to just use chilled water so that the whole drown mass of reaction mixture ends up at
about 30-35 C. At this temperature it is a heavy liquid and separates from the diluted acid easily. Tomorrow I will investigate the work up of the DCM
solution.
I will do a full write up then and if it is successful I will run a full scale preparation at about 0.5 moles.
|
|
|
clearly_not_atara
International Hazard
Posts: 2800
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
All electrophilic substitutions on fluorobenzene bias para.
https://pubs.acs.org/doi/10.1021/ed080p679
As such a high proportion of mononitro compound should be achieved by simply controlling stoichiometry, as all of the other positions are much less
reactive even from the beginning.
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Hi Boffis!
Believe it or not, yesterday I was surfing the web and came across the following patent. I found it interesting and saved. Who knows, someone might
find it usefull too. :-)
Enjoy - although you are finished the nitration process so I'm a bit late with this find!
Attachment: US4036838.pdf (1.2MB)
This file has been downloaded 335 times
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Thanks Pumukli! Interestingly I have ended up with just about the same process, presumably following the same logic. The only difference is that I am
trying to use nitrate salts and sulphuric acid as they are easier to obtain than 98% nitric acid for amateurs. But that patent is certainly
interesting and useful.
Ultimately, the ideal would be to separate the organic nitric acid solution from the sulphuric acid/sodium salt residue before use.
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Unfortunately DCM can only dissolve 1-2 % HNO3. (This is from one of the books in the SM Library, Energetic Materials "subsection". Read it years ago
so I can't be really sure.)
Yes, it is interesting that your method is closely mimics that of the patent. :-)
I plan to nitrate a chloro-benzoic acid soon hence I searched for the best method and found that patent. I still have my questions (separation of
isomers) though. :-)
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Pumukli, somewhere I am sure I have seen a preparation using nitric acid extracted into DCM and I calculated that the concentration must be about
0.12 molar, I have tried this morning to find the ref without success but I'll keep looking. Anyway its way too dilute for most practical uses without
concentration but the solubility should be enough to act as a contact improver. Below is my initial DCM mediated nitration experiment. No pics this
time I'm affraid but maybe on the next attempt.
Experiments into the mono-nitration of bromobenzene in a dichloromethane medium
Boffis 9/Dec/2020
This report covers my initial experiment into the nitration of bromobenzene in a dichloromethane medium using sulphuric acid and sodium nitrate as the
nitration agent. My initial attempts at nitrating bromobenzene were highly successful but required constant attention for 1.5 hours and towards the
end of nitration truly excruciating kneading of the reaction product to achieve full nitration. There has to be a better way!!
Industrially the reaction of both chloro and bromobenzene with nitrating mixtures are carried out using powerful propeller type mixing blades in
enclosed reactors that are way beyond the capability of laboratory scale preparations let alone amateur laboratory conditions. So I began to think
about ways to reduce the viscosity of the reaction mixture which can reach the consistency of cold butter by the end. Heat would help but has the
disadvantage that it quickly promotes dinitrations. My initial idea was to use anhydrous nitric acid in dichloromethane prepared by extraction a
solution of nitrate salt in conc. sulphuric acid with dichloromethane. The problem is that when looking into the process of extracting nitric acid
into DCM the maximum acid concentration at 70-80% recovery is only a little over 8% or a little over 0.1 molar. Thus to nitrate 25g of bromobenzene
(0.16 moles) would require roughly 1.5L of DCM. This did not seem to be a particularly practical option. On the other hand would it be possible to use
just a small amount of DCM as a medium to reduce the viscosity of the organic phase and to improve contact between the aqueous acid phase and the
organic phase? One further advantage of using dichloromethane in the reaction mixture is that it boils at 40°C which is just about the ideal
temperature for the mononitration of chlorobenzene, bromobenzene and p-dichlorobenzene.
The following experiment is an initial attempt to answer this question.
Experimental
14.2g (roughly 5% excess) of dry, finely ground sodium nitrate were mixed with 27ml of concentrated sulphuric acid (SG. 1.84) in a 150ml conical flask
on a stirrer hotplate and equipped with a magnetic stir-bar. A reflux condenser was added to the flask and then 10ml of dichloromethane were added
through it (Note 1). The mixture was stirrer for about 10 minutes but the sodium nitrate did not dissolve completely; fearing that this might result
in incomplete nitration I added 5ml of 70% nitric acid (0.08 moles so raising the excess to 55%), partly to increase the volume of aqueous phase and
partly to add some water in the hope that this would increase the solubility of the nitrate salt (Note 2).
25.00g of bromobenzene (0.16 moles) were mixed with 20ml of dichloromethane and added dropwise from a dropping funnel through the condenser to the
vigorously stirred acid mixture. The reaction mixture soon became warm and slightly yellow and then began to boil. The rate of addition was adjusted
so that the mixture refluxed very gently. The addition took about 20 minutes, once complete the stirring was continued for 1hr 10mins more. The
temperature soon began to fall so a little external heat was applied using the hotplate to maintain the temperature at reflux throughout the stirring.
The organic phase remained liquid and completely stirrable throughout.
When the reaction was complete the mixture was poured into a beaker containing 100g of ice and stirred manually until the ice had melted. The product
partly solidified so the volume was made up to 200ml with hot water; the organic phase quickly liquefied (Note 3). Most of the acid layer was decanted
off and the remaining liquid poured into a separating funnel were it was washed twice with 100ml portions of warm saturated sodium bicarbonate
solution and then twice with warm water. The organic layer was run into a beaker, covered and left to cool overnight at about 8°C (room
temperature!). The layer appeared to solidify into a pale yellow solid but when disturbed contained much oil. The solid was filtered off using a small
Buchner funnel (paper type), drained using the vacuum and washed with a little cold DCM. The almost white crystals weighed 9.470g and appear to be
mainly 4-nitrobromobenzene (29.5% of theory). The yellow filtrate was placed in a porcelain bowl on a hot water bath and heated to about 1.5 hours to
remove the DCM and any water present. The oil was allowed to cool and solidified to a soft butter-like solid weighing 16.970g. This was dissolve in
25ml of rectified spirit, in which it dissolved easily indicating very little 4-nitro isomer was present, and cooled. The yellow bladed crystals were
filtered off, washed with a very little cold rectified spirit and dried to give 6.128g of crude 2-nitro isomer (19% of theory). The filtrate contained
about 0.5ml of yellow oil which was removed (and preserved) before being evaporated down to about 12ml. On cooling no crystals formed only about 6ml
of pale yellow oil separated from the alcoholic liquor; its composition will be investigated TLC.
The 9.47g of crude 4-isomer was recrystallized from 60ml of rectified spirit but there was some dense fine grained salt present. This was allowed to
settle and the bulk of the clear supernatant solution poured into a fresh beaker where it rapidly crystallised (Note 4). The beaker was chilled to
4°, filtered, the cake washed with a little cold rectified spirit and dried to give 7.221g of almost white 4-nitro isomer. The filtrate was
evaporated down to about 25ml and cooled to yield a further crop of 0.304g of rather yellow crystals that require investigation.
Note 1) The small amount to dichloromethane added to the aqueous phase is to control the reflux temperature at the beginning of the addition.
Note 2) In the end the aqueous phase never became homogeneous but providing the sodium nitrate is very fine and well dispersed this may not be a
problem. It was originally intended that the acid mixture would be added to the organic phase but the lack of homogeneity meant that it would have
probably blocked the dropping funnel tap. I intend to investigate the use of azeotropic nitric acid for the mono-nitration later. It may also be
possible to use a concentrated sodium nitrate solution and extra sulphuric acid to ensure that the aqueous phase is homogeneous.
Note 3) If I run this reaction again I will add another 10ml or so of DCM to help maintain the organic phase liquid. The sodium bicarbonate wash
solution and water should be warmed to about 30-35°C too as this helps to prevent the formation of solids that block the tap in the separation
funnel.
Note 4) To avoid the problem of salt residues in the first crop of crystals it may be better the filter the warm, completely liquid oil and then cool
or even to filter and distil off the DCM to obtain a single crude product.
Discussion
The basic idea was a clear success but the recovery was rather low: only about 50% 2 and 4-nitrobromobenzene isomers plus about another 8g or so of
intractable oil (say 25%). The washings were not discarded immediately and over a day or so the small oily patches floating on the meniscus
solidified. These were filtered off to give another 1.38g of moist material or about 4% assuming it is nitrobromobenzene. It is assumed that the
remaining 20% was unreacted bromobenzene that volatilized along with the DCM and water on the water bath. If this is the case it may be better to
distil the oil to recover the volatiles but at this scale (0.16 moles) it wasn’t worth the effort.
More concerning is the overall level of nitration if the losses are due to much residual bromobenzene in the oil. This is particularly so as the
excess of nitrating agent was >50% i.e. one and a half time the amount required due to the addition of the extra nitric acid. The low temperature
probably limits the amount of di-nitration product and the presence of such compounds in quantity would have increased the apparent yield. This
suggests that there is a problem with the heterogeneous sodium nitrate /sulphuric acid, perhaps a layer of sodium bisulphate coats the residual sodium
nitrate preventing it from reacting. Interestingly, this does not appear to be the case in the solvent free nitration of p-dichlorobenzene where the
yield is almost quantitative with only a 3% excess of sodium nitrate but in this case the ration of acid to nitrate salt is much higher.
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Congratulations! Another good-quality work from you. 
I agree with the issues mentioned in the discussion. The level of nitration is unexpectedly low! I don't know what you have at hand but I'd surely
change sodium-nitrate to ammonium-nitrate. Perhaps the ammonium salt had a (much) better solubility and it'd help if that was the problem... Or maybe
some acetic acid as co-solvent?
Not to mention the reaction time. You finished in 20 minutes. What if you left it on stirring for an extra hour or two, without external heating (to
supress dinitration)? Yes, I know, it's easy to ponder on "what if"-s and it was just a one-shot experiment.
The isomer ratio is also interesting. I'd expected much higher para/ortho ratio, you got roughly p/o=1.5? Is it just me, my gut feelings or what do
you think about it? Did you expect such well-balanced p/o ratio?
I'd also be curious about the characterisation data of the products: melting points, tlc Rf values, the looks of the TLC plates, etc. You can do it!

[Edited on 10-12-2020 by Pumukli]
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
Boffis,
Nice peice of work. I agree with Pumukli that a longer reaction time may be useful. You should be able to follow the reaction by tlc to get some idea
of starting material disappearance. I look forward to further reports.
AvB
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi guys, thanks for the comments, The 20 minutes was just the addition time. The reaction was then stirred for a further 70 minutes to give 1.5 hours
total which is the same as the reaction time used in the kneading process.
It is interesting that the recoved p to o ratio of the original preparation (no DCM) was about 2.5 to 1 were as with DCM it appears to be as you say
1.5 to 1. It is also interesting that no p- isomer could be reacoved from the oil after the removal of the first crop of p isomer. This suggests that
DCM may be a better solvent for the separation of the o and p isomers than rectified spirit.
I am about to run some TLCs on the various products and fraction now I have some micro pipetters and tips (tips arrived yesterday care of Ebay!) I like your idea Pumukli of an additional co-solvent like acetic acid. In fact I
am wondering if I could ensure of homogeneous nitration mixture by making up a warm concentrated NaNO3 solution in water and slowly adding ice cold
sulphuric acid and using rather more acid than strictly required to act as a dehydrating agent with possibly some acetic anhydride to control the
water activity. It would be easier and safer if I could dissolve the NaNO3 in glac. acetic acid first rather than water. Any idea on the solubility of
NaNO3 in acetic acid?
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Well I have run a series of TLCs of the various product from the above experiment and the results are interesting. In my original nitration
experiments I used pure 100-120 Bp range petroleum ether to develop the chromatographs. In these recent one I have done as was suggested in the
dichlorobenzene nitration thread and used a mixture of 8:1 petroleum ether Bp R 60-80C:ethyl acetate. This does seem to have worked, increasing the
resolution significantly. Using a micropipetter (5ul) helps get more consisted spots (I am going to reduce the spots to 3ul in future).
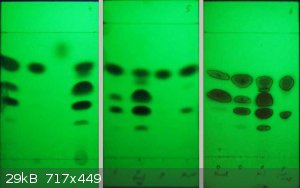
The 3 TLC plates are No.s 4,5 and 6 from left to right and reveal:
Pl4-Ch1 Crude crop of "o" isomer, interestingly the plate reveals that it is mainly 2,4-dinitro and 4-nitro with minor 2-nito
Pl4-Ch2 1st crop recrystallised "p" isomer =almost pure
Pl4-Ch3 Bromobenzene starting material rather volatile
Pl4-Ch4 "p" alcoholic residue after 2 crops of crystals
Pl5-Ch1 Crude "o" as in PL4-Ch1
Pl5-Ch2 final oil from "o" isomer recovery
Pl5-Ch3 "p" 1st crop recrystallised as in Pl4-Ch2
Pl5-Ch4 "p" 2nd crop of crystals
Pl6-Ch1 "o" isomer recystallised from earlier experiments
Pl6-Ch2 "o" isomer as in PL4-Ch1
Pl6-Ch3 "o" isomer final oil as in Pl5-Ch2
Pl6-Ch4 "p" isomer 2nd crop of crystals =Pl5-Ch4
From these TLC it follows that the initial 9.47g of product recovered from the DCM oil is mainly 4-nitrobromobenzene and the 7.221g of
recrystallisation 1st crop is essentially pure 4-nitrobromobenzene (Pl4-Ch2). The second crop is noticably contaninated but not with
2-nitrobromobenzene, although I can't be certain at this stage, the impurity, based on the earlier nitration experiment, is probably
2,4-dinitrobromobenzene. The alcoholic residue left after the second crop of crystals (Pl4-Ch4) contains 2-isomer > 4-isomere = 2,4-dinitro isomer
and a little of what is probably 2,6-dinitro-isomer.
Processing of the residue left after the water bath removal of the DCM and recrystallisation of rectified spirit gave 6.128g of the "o"-isomer
(Pl4-CH1 and Pl5-Ch1). Inspite of its distinct yellow colour the 1st crop of crystals appears to be 4-nitro > 2-nitro and even 2,4-dinitro >
2-nitro! A further small crop of yellow crystals c 1g was recovered by evaporating the filtrate down by half (not TLC'ed) giving a final yellow oil
that appears to be mainly 2-nitro and 2,4 dinitrobromobenzene with minor 4-nitro and traces of 3-nitro and 2,6-dinitro. It is hardly surprising that
the Mp of the final oil is <10 C!
The Rf values vary a bit from TLC to TLC but when normalised to the 4-nitrobromobenzene =0.74 they are remarkably constant.
Bromobenzene lies at about 0.88 but is highly effemoral, disappearing within 30 minutes.
Possible 3-nitro isomer at 0.68
2-nitro isomer at 0.57
2,4-dinitro isomer at 0.44
possibly the 2,6-dinitro isomer at 0.29
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I have just completed a second nitration of bromobenzene on twice the scale. This time I warmed the nitrating mixture until it was a clear liquid and
added it to 50.00g of bromobenzene in 50ml of DCM. Using only a magnetic stirrer and only 3% excess of nitrating agent the nitration worked well but I
found that temperature control of everything is vital and more DCM had to be added to the mixture to keep the oil liquid during washing. The overal
yield of crude product was 62.67g, assuming that the only mononitro compounds were formed then the yield was 96.8% of theory. When the DCM solution
was finally cooled it deposited 19.88g of crude white p-isomer dominated material. The DCM was distilled off from the filtrate and the oil allowed to
solidify to give 42.39g of crude "mixed" isomers. I have yet to run TLC on the products.
I was wondering too about separating the mixture with a silica column, I have the material to make such a column but the question is how do I know
where to cut the fraction? Can this be calculated from the the Rf value if I use the same solvent mixture as for the TLCs?
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Quote: Originally posted by Boffis  |
I was wondering too about separating the mixture with a silica column, I have the material to make such a column but the question is how do I know
where to cut the fraction? Can this be calculated from the the Rf value if I use the same solvent mixture as for the TLCs? |
The Rf in column chromatography will be similar to the Rf in TLC but not exactly the same.
One way, that works pretty well in my experience, is to collect a bunch of small fractions, then run TLC on each one and figure out which ones contain
each compound. You can then combine those fractions.
[Edited on 2020-12-14 by Metacelsus]
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Nice work. As I can see you are working hard in these days it is an inspiration for me to do something meaningful as well. We'll see.
Regarding the chromatography wouldn't it be possible to check the effluent from the column continuously say with an UV lamp? Maybe these coumpounds
would show up in that invisible light? The o-isomer is yellow, isn't it? Then you might be able to see when it arrives. Just thinking aloud.
Or, instead of running dozens of spots on a TLC, what if you took a drop from each fraction on a stray piece of fluorescent TLC plate, quickly dry it,
then put it under UV and see if there was anything that quenched the fluorescence or not. If not: you could reuse the TLC snippet again. I never tried
such a ghetto method but I think it could work and would be quick...
I also think that purifying something on the 40 g scale requires a decent (wide) column, lots of silica and even more solvent... And lots of glassware
to catch those fractions.
If you give it a go please don't forget to take some pictures!
[Edited on 14-12-2020 by Pumukli]
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Thanks for the ideas guys. I will have a look at these over the next day or so. I do have a lot of small 5 and 10ml beakers so I may be able to do an
incremental collect like metacelsus suggests and then TLC them. My columns are only about 18 to 19mm ID so the loadingis not going to be more than a
few gram ata time.
I have just run a TLC on the second DCM mediated nitration. The results are much cleaner than before much less dinitro stuff.
The initial crop of crystals from the cooled DCM nitration solution was found to consist mainly of the 4-isomer as expected though traces of the 3-
and possibly the 2,4-dinitro isomer were just detectable. Residue after removal of the DCM from the filtrate contains mainly the 2-isomer, a smaller
but significant amount of the 4 isomer and a little 2,4-dinitro compound. These results are better than the previous experiment because the nitrating
mixture was added to the bromobenzene so the later was in excess until the very end so less dinitro products were generated and the homogeneous
nitrating mixture was present in only 3% excess which also help prevent over nitration and oxidation. The product does not give an orange colour with
sodium carbonate suggesting less oxidation.
My plan now is to use the fractional crystallisation method I developed before to get the 4 and 2 nitrobromobenzene isomer pure and then try column
chromatography on the intractable oil. I think the colours are too faint to use for the separation of the fractions so I may try lots of small beakers
and tlc spots.
|
|
|








