| Pages:
1
2 |
michalJenco
Hazard to Self

Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
Cyclic diester from 2,2'-oxydibenzoic acid?
Hi fellow chemists.
I recently made 2,2'-oxydibenzoic acid (btw check that fucked up image on SigmaAldrich  ). ).
Looking at the structure, could a double fischer esterification happen with a diol like ethylene glycol or longer? I wasn't able to find any
reference. I don't see any reason why it shouldn't happen, but maybe some of you have actual experience.
I'm thinking something like this:

Edit: anyone try to name the product? My knowledge of naming is not enough here.
[Edited on 14-6-2020 by michalJenco]
|
|
|
DraconicAcid
International Hazard
Posts: 4357
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
No idea, but how did you make the acid?
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
I did condensation of salicylic acid with o-chlorobenzoic acid in very basic very hot water.
Edit: I also read it can be made by that procedure from the o-chlorobenzoic acid alone, but to me my method felt that it would consume much more of
the chlorinated acid, which I don't want sticking around.
[Edited on 14-6-2020 by michalJenco]
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
You're trying to make an 11-membered ring which is really unfavorable, but perhaps the structure is twisted in a way that makes it less terrible.
|
|
|
Syn the Sizer
National Hazard
Posts: 600
Registered: 12-11-2019
Location: Canada
Member Is Offline
|
|
Quote: Originally posted by UC235  | | You're trying to make an 11-membered ring which is really unfavorable, but perhaps the structure is twisted in a way that makes it less terrible.
|
an 11 membered carbon ring is very unfavourable, but I think with the 3 oxygens would would reduce strain on the carbons.
Edit:
That is an odd depiction on the Sigma site.
[Edited on 15-6-2020 by Syn the Sizer]
|
|
|
Cou
National Hazard
Posts: 958
Registered: 16-5-2013
Member Is Offline
Mood: Mad Scientist
|
|

Interesting molecule, but I predict this might not work b/c of tar formation. once the ethylene glycol forms an ester with one carboxylic acid... the
other alcohol group on the ethylene glycol could form ester with the carboxylic acid of another molecule.
I'm brainstorming ideas here. esterification with 1 molar equivalent of 2-bromoethanol will give a mixture of diester, monoester, and no ester
products. You could use fractional distillation to isolate the monoester, if the boiling points of all 3 are different enough. If the monoester is
diluted in a large volume of organic base such as pyridine (to minimize intermolecular reactions), an intramolecular SN2 reaction could happen.

|
|
|
Sigmatropic
Hazard to Others
Posts: 307
Registered: 29-1-2017
Member Is Offline
Mood: No Mood
|
|
If you could dehydrate it to the cyclic anhydride then that would be a nice access point to the bromoethyl monoester.
|
|
|
Pumukli
National Hazard
Posts: 708
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Have you checked the price of the acid what you've just synthetized? :-)
Sigma wants a pleasant pile of money for a mere 25 mg (!) :-)
The demand may be low though... :-( Otherwise what a lucrative (and legal) business we may miss! :-)
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Sigma-Aldrich pricing is bonkers. There's no way that's actually worth $5560/g.
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I would be interested in the procedure you used to prepare the acid. Can you give us a reference or some details of what you did? Very interesting
stuff.
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
| Quote: Originally posted by Pumukli | Have you checked the price of the acid what you've just synthetized? :-)
Sigma wants a pleasant pile of money for a mere 25 mg (!) :-)
The demand may be low though... :-( Otherwise what a lucrative (and legal) business we may miss! :-) |
I have checked the price and it really is bonkers, considering how trivial the precursors and synthesis are. I tend to view these prices as
signatures of non-existent demand as anyone who will search for that specific chemical is definitely capable of synthesizing it themselves I previously made 40g of this stuff (4-methyldaphnetin) - worth roughly $280,000 - so that thinking is the only way to sleep at night for me.
Edit: Or, you know, maybe I didn't make what I think I did and it actually is harder to make and the price justified. I will write a full write-up in
this thread (as someone is interested) later so you can judge my procedure.
[Edited on 15-6-2020 by michalJenco]
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
| Quote: Originally posted by UC235 | | You're trying to make an 11-membered ring which is really unfavorable, but perhaps the structure is twisted in a way that makes it less terrible.
|
Yes that's what I'm thinking .. repulsion of the rings could squish the carboxyls closer to each other. But I have no idea how much certain bonds can
twist, it looks favorable because of the particular way I drew it originally.
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
| Quote: Originally posted by Cou |
Interesting molecule, but I predict this might not work b/c of tar formation. once the ethylene glycol forms an ester with one carboxylic acid... the
other alcohol group on the ethylene glycol could form ester with the carboxylic acid of another molecule.
I'm brainstorming ideas here. esterification with 1 molar equivalent of 2-bromoethanol will give a mixture of diester, monoester, and no ester
products. You could use fractional distillation to isolate the monoester, if the boiling points of all 3 are different enough. If the monoester is
diluted in a large volume of organic base such as pyridine (to minimize intermolecular reactions), an intramolecular SN2 reaction could happen.
|
Yeah I presumed tar would form as there is no reason ethylene glycol would not connect two separate reactant molecules. It might even make some amount
of this monster, analogous to the side product of reaction of catechol and diiodimethane:
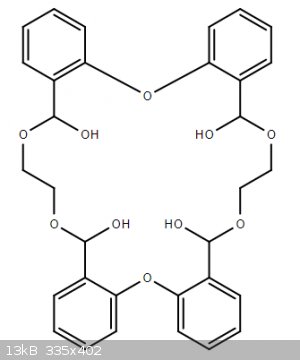
I love that you actually thought about it and proposed a mechanism, I didn't know about the last organic base-catalysed mechanism. I really appreciate
that! Unfortunately I don't have access to 2-bromoethanol and its synthesis is beyond my current capabilities. This will be a great thread to come
back to if I do.
The official name will haunt me.
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
The cyclic anhydride would be really interesting by itself! I'll have to look into that because I never made an anhydride.
|
|
|
Cou
National Hazard
Posts: 958
Registered: 16-5-2013
Member Is Offline
Mood: Mad Scientist
|
|
In that case, maybe you could just react ethylene glycol with the anhydride to get a mono ester, then intramolecular esterification after dilution to
minimize intermolecular reaction.
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
| Quote: Originally posted by Boffis | | I would be interested in the procedure you used to prepare the acid. Can you give us a reference or some details of what you did? Very interesting
stuff. |
Synthesis of 2,2'-oxydibenzoic acid
I think of this synthesis as a Williamson ether synthesis. I am not educated as a chemist so I piece things together from the internet. Similar
reactions are described, though (e.g. salicylaldehyde + chloroacetic acid on the Williamson ether synthesis wiki, which I did and it worked), so I work assuming the principle is general. In general, you want enough base to
depronate all carboxyl and phenol groups in all your reactants and then an excess to achieve a very basic mixture even with everything deprotonated. I
used 1.5x molar here.
Materials:
KOH - 54.8g (977mM)
Salicylic acid - 30g (217mM)
2-chlorobenzoic acid - 34g (217mM)
35% Hydrochloric acid - as needed
Water - 160g + enough to get to ~430mL of reaction mixture
Procedure:
Magnetic stirring on a hotplate is implied throughout the reaction, heating starts off. Fits neatly into a 500mL erlenmeyer flask. Solid stirring is
required as a lot of precipitate has to be stirred.
Dissolve the base in the first portion of water.
When clear solution is reached, add B which dissolves quickly.
Wait for 5 minutes and add C which also dissolves quickly.
Turn on heating and bring the reaction mixture right below its boiling point (around 118 °C) and hold it there for two hours.
Turn off heating and add the other portion of room temperature water (temperature of reaction drops to ~80 °C) and acidify with D until pH 0-1.
A substantial of insoluble solids are formed - our product.
Turn on heating and bring near boiling point and keep just under it for 10 minutes.
This makes a lot of the product actually dissolve, and the undissolved part melts into a bottom golden layer (droplets while stirring but they settle
into a layer when stopped).
Stop the heating and let the reaction mixture cool while stirring until the product is no longer melted, then you can turn off stirring.
The whole flask fills with plate-like pure white crystals.
If left unstirred while cooling, a lot of the product collects in a bottom layer which, when left to solidify, is very hard to break up and get out of
the flask.
Workup as usual - vacuum filter, wash with water until filter water is not acidic.
The wet product can be recrystallised as such, no need to dry.
I recrystallised from about 350mL of 1:3 (volume, roughly) EtOH:water, but I would use even less EtOH because product is extremely soluble in it even
at room temperature. The amount of product from this synthesis could be recrystallised entirely from like a liter of water, I think.
Wash with cold water after recrystallisation. Let the white fluff dry on air for a few days.
The yield is 52.8g (204mM, 94.2%) of pure white powder-like mass of tiny crystals. Faintest or no smell at all.
Rationale:
I have done several similar reactions between ortho and para chlorobenzoic acids acid phenols, because the procedure is simple, I
have access to both of these groups of chemicals and I am curious. I condensed both phenol and hydroquinone with both o- and
p-chlorobenzoic acid to make the following products:
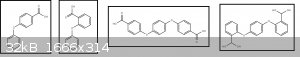
The underlying motivation is my curiosity for smells (of esters), so I made methyl esters of all of those 4 molecules. They have a beautiful smell.
The 2- substituted esters smell very similar to methyl salicylate, while the 4- substituted ones are more similar to
4-acetphenetole.
For the sake of interest - methyl esters of the first three are solids while the 4th is actually a liquid, which surprised me a lot.
[Edited on 15-6-2020 by michalJenco]
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
| Quote: Originally posted by Cou |
In that case, maybe you could just react ethylene glycol with the anhydride to get a mono ester, then intramolecular esterification after dilution to
minimize intermolecular reaction. |
This actually looks like a viable way for me to do it. The separation of anhydride will be pretty challenging I think. Don't have vacuum.
It also avoids brominated alcohols, which is very welcome, great suggestion!
[Edited on 15-6-2020 by michalJenco]
|
|
|
Cou
National Hazard
Posts: 958
Registered: 16-5-2013
Member Is Offline
Mood: Mad Scientist
|
|
What does 2,2'-oxydibenzoic acid smell like?
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Cou |
but I predict this might not work b/c of tar formation. once the ethylene glycol forms an ester with one carboxylic acid... the other alcohol group on
the ethylene glycol could form ester with the carboxylic acid of another molecule. |
Why should it take the
trouble to react with another molecule when it has a COOH right in front of its face!
I suggest you slowly drip ethylene glycol into a pot containing your diacid and TCT.By keeping the concentration of the smaller(and more reactive)
component low ,you can minimise the chances of side reactions
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
It has a very faint smell but not specific enough to describe. I just smell something something as opposed to completely nothing. Definitely not an
interesting or a pleasant smell, though.
Its dimethyl ester, though, a different animal. Wonderful smell almost identical to methyl salicylate, but with a slight twist. I may be imagining the
twist because I know it is something different, though.
Edit - wrote benzoate instead of salicylate
[Edited on 16-6-2020 by michalJenco]
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
| Quote: Originally posted by CuReUS | | Quote: Originally posted by Cou |
but I predict this might not work b/c of tar formation. once the ethylene glycol forms an ester with one carboxylic acid... the other alcohol group on
the ethylene glycol could form ester with the carboxylic acid of another molecule. |
Why should it take the
trouble to react with another molecule when it has a COOH right in front of its face!
I suggest you slowly drip ethylene glycol into a pot containing your diacid and TCT.By keeping the concentration of the smaller(and more reactive)
component low ,you can minimise the chances of side reactions |
Well, it has it right in its face the way I drew the diagram. It's just as possible for the benzoic acid groups to be rotated opposite to each other
on the oxygen atom, right?
TCT is cyanuric chloride? No way for me to:
Get
Want to work with
Are there any alternatives? This is an area of chemistry that is opaque to me at this time.
|
|
|
myr
Harmless
Posts: 48
Registered: 18-7-2018
Member Is Offline
|
|
| Quote: Originally posted by michalJenco | | Quote: Originally posted by Boffis | | I would be interested in the procedure you used to prepare the acid. Can you give us a reference or some details of what you did? Very interesting
stuff. |
Synthesis of 2,2'-oxydibenzoic acid
I think of this synthesis as a Williamson ether synthesis. I am not educated as a chemist so I piece things together from the internet. Similar
reactions are described, though (e.g. salicylaldehyde + chloroacetic acid on the Williamson ether synthesis wiki, which I did and it worked), so I work assuming the principle is general. In general, you want enough base to
depronate all carboxyl and phenol groups in all your reactants and then an excess to achieve a very basic mixture even with everything deprotonated. I
used 1.5x molar here.
Materials:
KOH - 54.8g (977mM)
Salicylic acid - 30g (217mM)
2-chlorobenzoic acid - 34g (217mM)
35% Hydrochloric acid - as needed
Water - 160g + enough to get to ~430mL of reaction mixture
Procedure:
Magnetic stirring on a hotplate is implied throughout the reaction, heating starts off. Fits neatly into a 500mL erlenmeyer flask. Solid stirring is
required as a lot of precipitate has to be stirred.
Dissolve the base in the first portion of water.
When clear solution is reached, add B which dissolves quickly.
Wait for 5 minutes and add C which also dissolves quickly.
Turn on heating and bring the reaction mixture right below its boiling point (around 118 °C) and hold it there for two hours.
Turn off heating and add the other portion of room temperature water (temperature of reaction drops to ~80 °C) and acidify with D until pH 0-1.
A substantial of insoluble solids are formed - our product.
Turn on heating and bring near boiling point and keep just under it for 10 minutes.
This makes a lot of the product actually dissolve, and the undissolved part melts into a bottom golden layer (droplets while stirring but they settle
into a layer when stopped).
Stop the heating and let the reaction mixture cool while stirring until the product is no longer melted, then you can turn off stirring.
The whole flask fills with plate-like pure white crystals.
If left unstirred while cooling, a lot of the product collects in a bottom layer which, when left to solidify, is very hard to break up and get out of
the flask.
Workup as usual - vacuum filter, wash with water until filter water is not acidic.
The wet product can be recrystallised as such, no need to dry.
I recrystallised from about 350mL of 1:3 (volume, roughly) EtOH:water, but I would use even less EtOH because product is extremely soluble in it even
at room temperature. The amount of product from this synthesis could be recrystallised entirely from like a liter of water, I think.
Wash with cold water after recrystallisation. Let the white fluff dry on air for a few days.
The yield is 52.8g (204mM, 94.2%) of pure white powder-like mass of tiny crystals. Faintest or no smell at all.
Rationale:
I have done several similar reactions between ortho and para chlorobenzoic acids acid phenols, because the procedure is simple, I
have access to both of these groups of chemicals and I am curious. I condensed both phenol and hydroquinone with both o- and
p-chlorobenzoic acid to make the following products:
The underlying motivation is my curiosity for smells (of esters), so I made methyl esters of all of those 4 molecules. They have a beautiful smell.
The 2- substituted esters smell very similar to methyl salicylate, while the 4- substituted ones are more similar to
4-acetphenetole.
For the sake of interest - methyl esters of the first three are solids while the 4th is actually a liquid, which surprised me a lot.
[Edited on 15-6-2020 by michalJenco] |
I don't think you formed the diaryl ether. Aryl halides do not normally undergo Williamson ether synthesis. Have you taken a melting point, or used a
test to determine how much (or if any) phenolic contamination is in the product?
|
|
|
Boffis
International Hazard
Posts: 1879
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@michaljemco; Thankyou for posting details of your prep.
@myr; how different is this reaction to the preparation of N-Phenylanthranilic acid (See Vogel, Practical Organic Chemistry, 3rd, page 991) from
aniline and o-chlorobenzoic acid? Except that the preparation in Vogel uses a copper oxide catalyst? Would a copper or copper oxide catalyst help in
this reaction?
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
Hmm that is worrying. I didn't do a m.p. test on the bottled product but it was liquid in the reaction mixture until around 80 °C. Salicylic acid has
a m.p. of 158 °C and 2-chlorobenzoic acid melts at 142 °C, so I definitely made something.
I just did a FeCl3 test and I got an intense purple coloration immediately. Looks like I failed miserably  . .
But I wonder. Molbase suggests you can make my product from the chloroacid alone. "Surely" throwing in a deprotonated salicylic acid should make it even better?
|
|
|
michalJenco
Hazard to Self
Posts: 50
Registered: 7-2-2019
Member Is Offline
|
|
This also makes me wonder about what I actually made when I did this procedure with phenol and 2- and 4- chlorobenzoic acids, where the products after
Fischer esterification with methanol were solids with amazing smells. Methyl 2-chlorobenzoate is a liquid so I didn't make that. I also don't think a
2-chlorobenzoate ester would smell exactly like salicylate with a twist, which my presumed methyl 2-phenoxybenzoate (2-PB) absolutely does
smell like. They smell exactly how you would imagine them based on the molecule - my presumed 2-PB is spicy like salicylate and 4-PB is somehow round
and sweet, anisic, like 4-acetphenetole (that I coincidentally also made, which makes me able to compare).
Also, I made methylparaben a while ago and it has no smell at all, so my 4-chlorobenzoic acid after reaction with phenol and base didn't turn (at least not
fully) into a 4-hydroxybenzoic acid. This is proof to me that some - maybe a lot - of my product actually is the diaryl ether carboxylic acid in these
types of reactions.
Also also the pungent smell of the chloroacids disappears during the reaction, so the chlorine is leaving the benzene ring. If it is leaving
and it is not leaving a hydroxy group behind at least some of the time, what other options besides diaryl ether are there?
Yeah, my only analytical instruments are my senses but you see that the evidence does seem to stack up.
[Edited on 17-6-2020 by michalJenco]
|
|
|
| Pages:
1
2 |








